A Hypothetical Mechanism of how HPV E6 and E7 Infections inactivate BRCA1 Function Resulting in Cervical Cancer
Chataigne Michara1, Nicholas Wilson1, Omojolaade Akintade1, Jingyao Xu1, Lauren Gibbs1, Okorie Ijeoima1, Lee S. Caplan2, E. Shyam P. Reddy1, Veena N. Rao1*
1 Cancer Biology Program, Department of OB/GYN, Atlanta, GA 30303, USA.
2 Community Health and Preventive Medicine, Morehouse School of Medicine, Atlanta, GA 30303, USA.
*Corresponding Author
Veena N. Rao,
Professor and Co-Director Cancer Biology Program, GCC Distinguished Cancer Scholar, Department of OB/GYN, Morehouse School of Medicine, RW D-335, 720 Westview
Drive, Atlanta, Georgia 30310-1495.
Tel: 404-756-5755, Fax 404-756-8828
E-mail: vrao@msm.edu
Received: January 24, 2024; Accepted: February 08, 2024; Published: February 16, 2024
Citation: Chataigne Michara, Nicholas Wilson, Omojolaade Akintade, Jingyao Xu, Lauren Gibbs, Veena N. Rao, et al., A Hypothetical Mechanism of how HPV E6 and E7 Infections
inactivate BRCA1 Function Resulting in Cervical Cance. Int J Chronic Dis Ther. 2024;9(1):137-140.
Copyright: Veena N. Rao�2024. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
Abstract
Cervical cancer is one of the most common female cancers,withHuman Papilloma Virus (HPV)infection being the major risk factor. Although regular screening for cervical cancer has been associated with reduced incidence of the disease, it is still critical to investigate how the cancer develops, so that we can improve treatment options. Previously, HPV E6 and E7 oncoproteins were shown to interact with BRCA1 to inactivate its functions but how this may initiate cervical cancer was unknown.Ubc9, p16 INK4 expression is strongly upregulated and ER- is declined in cervical lesions. We and others observed a similar scenario in triple negative breast cancer (TNBC) with BRCA1 mutation or dysfunction where loss of binding to its downstream target Ubc9 results in ER-a repression and high p16INK4expression. We therefore hypothesize that HPV E6 E7 oncoproteins by tethering BRCA1 inactivates itsfunction causing loss of binding to Ubc9 resulting in its accumulation, ER-a downregulation, high p16INK4 levels resulting in cervical cancer. In summary, wehave proposed a novel hypothetical molecular mechanism as to how HPV oncogenes interfere with BRCA1 function contributing to the development of cervical cancer. If confirmed future work will help in using Ubc9 and its downstream targets as potential biomarkers for early diagnosis and/ or monitoring the progression of HPV oncogenic infections and for designing drugs that target Ubc9 expression to combat these aggressive cancers.
2.Introduction
7.Conclusion
5.References
Keywords
Cervical Cancer; HPV E6 E7; BRCA1; Ubc9; ER-a; p16 INK4; etc.
Abbreviations
HPV: Human papillomavirus, TNBC: Triple Negative Breast Cancer; ER: Estrogen Receptor; AA: African
American;Ubc9: SUMO E2- conjugating enzyme 9.
Introduction
HPV and Cervical Cancer
Cervical cancer is one of the most common cancers in women,
despite a decline in both incidence and mortality rates over the
past 43 years (1975-2018), with 14,100 estimated new cases and
4,290 estimated deaths in 2022 [1]. Hispanic women have a higher
incidence of cervical cancer than non-Hispanic Black and White
women, while non-Hispanic Black women have a higher mortality
rate than Hispanic and non-Hispanic White women [1]. Regular
screening for cervical cancer has strongly reduced cervical
cancer incidence, as it enables us to identify and eliminate early
neoplasms. Human Papilloma Virus (HPV) is the most common
risk factor for cervical cancer, with HPV DNA found in 99% of
invasive cervical cancers worldwide [2, 3]. Most cervical cancers
involve the interactions between HPV oncogenes E6 and E7 with
tumor suppressor genes like p53 and pRB promoting their degradation
[4]. Focusing on these interactions, we have proposed a
novel mechanism by which E6 and E7 oncogenes cause cervical
cancer via inactivation of BRCA1 function. BRCA1 and BRCA2
are tumor suppressor genes essential for the repair of damaged
DNA,and mutations in these genes increase the risk of developing several types of cancers,most notably breast and ovarian
cancers. However, studies have shown that BRCA1 and BRCA2
gene mutations can also increase a woman�s risk of developing
cervical, uterine, pancreatic, colon, stomach, esophageal, and liver
cancers [2].
BRCA1 and Cervical Cancers
BRCA1 and its isoformsfunctionas growth / tumor suppressorsin
TNBC, ovarian and prostate cancer cells and mice xenografts
[5]. In normal cells, BRCA1/1a/1b proteins bind to adownstream
target Ubc9 to facilitate nuclear localization of BRCA1, resulting
in the activation of ER- a BRCA1 mutations/dysfunctiondisrupt
this normal processleading to loss of binding to Ubc9, resulting
in high levels of Ubc9 which enters the nucleus and suppresses
ER- a,causingTNBC [6, 7].
A similar scenario is seen in cervical cancers where Ubc9 is
strongly upregulated, increase in p16INK4 expression and ERato
be strongly repressed in cervical cancer lesions [8, 9]. HPV E6
and E7 oncoproteins have been shown to inactivate BRCA1 function
[8] in cell lines (siHA, Caski and HeLa).An inverse relationship
is observed between ER- a and p16 INK4 in cervical cancer
tissue, suggesting that HPV infection can cause loss of ER- a
expression and increase in P16 INK4 as the cells become cancerous,
p16 INK4 is a marker for HPV linked cancers [9]. Based on
these observations we put forth a novel molecular mechanism of
inactivation of BRCA1 by HPV E6/E7 infectionresults in loss of
binding to Ubc9 which at high levels, represses ER- a and increases
p16 INK4 activity, apoptosis inhibition by inactive cytoplasmic
BRCA1 resulting in cervical cancer.
HPV on coproteins are known to interact with p53 to upregulate
Ubc9. Specifically, the E6 oncoprotein can form a complex with
p53 and a ubiquitination enzyme E6-AP [10]. By binding and degrading
p53, cell cycle checkpoints can be bypassed, and tumor
cell progression can be enabled. The E7 oncoprotein on the other
hand binds to the Rb domain that is responsible for tumor suppression.
Normally, Rb binds to E2F-family transcription factors
to regulate the cell cycle. When the E7 oncoprotein binds to Rb
to disrupt its interaction with E2F, E2F factors are released in
their active forms, and the cell cycle can be constitutively active,
thus allowing for tumor formation. During cancer formation and
progression, oncoproteins HPV E6 and E7 can concurrently bind
to cell cycle mediators p53 and Rb, as well as BRCA1 [8]. In binding
to BRCA1, HPV E6 and E7 bind to amino acids 67-100 and
1532-1749 in the C-terminal domain of BRCA1. As opposed to
the degradation seen in p53 and Rb upon binding to the oncoproteins,
BRCA1 is not degraded but loses its function upon binding
to HPV E6 and E7 [8]. Previously a new BRCA1-Ubc9 nuclear
trafficking pathway was identified and BRCAness was found to
perturb this balance resulting in TNBC [11].
HPV- positive Cervical Cancers and Ubc9
Sumoylation, the post-translational modification process of adding
a small ubiquitin-like modifier or SUMO moiety to proteins,
is mediated by the E2-conjugating enzyme Ubc9 [12]. As such,
Ubc9 levels are implicated in an array of cellular functions by
interacting with and regulating cell cycle proteins and tumor suppressors
[13, 14]. A proposed mechanism of Ubc9-related tumorigenesis
has been documented in the literature, with high levels of
Ubc9 implicated in cancers [15]. Though not exhaustive, a few examples
of Ubc9�s involvement in cancer include its role in breast
cancer metastasis and tumor cell invasion, as well as its involvement
in cervical cancer tumor progression [16, 17]. Experiments
carried out by Mattoscio et al. demonstrated that HPV-positive
cervical cancer lesions displayed higher Ubc9 levels compared to
HPV-negative head and neck cancer lesions; results also demonstrated
an upregulation of Ubc9 correlating with cervical cancer
lesion progression [18]. It was speculated that the upregulation
of Ubc9 was promoted by the E6 and E7 oncoproteins via p53.
This upregulation prevents cell apoptosis. By itself, Ubc9 reduces
apoptosis, while the E6 and E7-induced transformation leads
to apoptosis resistance [15, 18]. Reduction of apoptosis in cells
would lead to a higher likelihood of tumorigenesis. This implies
that HPV E6 and E7 oncoproteins inhibit Ubc9 degradation, thus
allowing Ubc9 accumulation as well as apoptosis-resistance in the
cell. Thus, Ubc9 detection can be used as a biomarker in diagnosing
and/or monitoring the progression of HPV oncogenic
infections [18].
P16 INK4 loss rescues BRCA1 Function:
BRCA1 is a known tumor suppressor that is involved in several
pathways of DNA repair [19, 20]. Cao et al., demonstrated that a
lack of BRCA1 caused premature senescence in cultured cells as
well as tumorigenesis in mice [21]. The same study found that this
malignant transformation of cells was mediated by p53 [21]. Interestingly,
Schuyer and Burns found that BRCA1-related breast
cancers contained a higher amount of p53 mutations relative to
sporadic cancers [22].
p16INK4a (p16) is a cyclin-dependent kinase inhibitor that limits
the progression of the cell cycle from G1 to S [23]. This kinase inhibitor
has been extensively studied and reported in the literature
as a biomarker for cervical cancer [24-26]. Khleif et al.,[23] found
an inverse relationship between p16INK4a and retinoblastoma
tumor suppressor protein (RB) in which mutated, deleted, or inactivated
RB led to higher expression of p16INK4a protein [23]. As
previously mentioned, HPV E7 binds and inactivates RB,causing
the release of E2F factors. Interestingly, Farzanehpour et al. demonstrated
a direct relationship between the overexpression of
p16INK4a and the severity of cervical cancer [27]. Moreover,
Lau et al [28] found that cells transfected with p16INK4a small
interfering RNA (siRNA) had a much higher rate of apoptosis
when exposed to ultraviolet irradiation and cisplatin compared to
the siRNA control counterpart, thus suggesting that p16INK4a is
implicated in the cellular apoptosis response [28]. Lastly, BRCA1
also interacts with Nrf2 to regulate cell survival [29].BRCA1 loss
or inactivation has been implicated as a cause of tumorigenesis in
the literature [30, 31]. Its role in tumorigenesis lies in its regulation
of DNA damage checkpoints. Loss or inactivation of BRCA1
results in premature senescence and apoptosis-resistant cells [31].
Scott et al. [32] further demonstrated that disruption of BRCA1
function could lead to premature senescence in mammary epithelial
cells [32]. However, the same study also found that loss
of p16INK4 rescued BRCA1 from loss of function by reducing
senescence in the mammary epithelial cells. This finding suggests
that p16INK4 is involved in DNA damage repair and cell senescence
and could be a potential downstream target in the BRCA1
pathway.
Clinical implications: HPV oncogenes [E6, E7] are known to play a vital role in malignant transformation. E6 oncoproteins
target cellular tumor suppressor protein p53 for inactivation and
degradation [33]. Likewise, E7 oncoproteins bind to RB, disrupting
its interaction with E2F, leading to uncontrolled tumor replication
[33]. The interactions between E6 and p53 as well as E7
and RB impair Ubc9 degradation and cause accumulation of it in
cells and tissues which increases host cell resistance to apoptosis
[34]. These interactions can be targeted for therapeutic interventions
to regain function of the tumor suppressor genes [33]. Also,
disrupting the interaction between BRCA1 and the E6/E7 oncoproteins
is another viable way that can be explored for treating
cervical cancers.
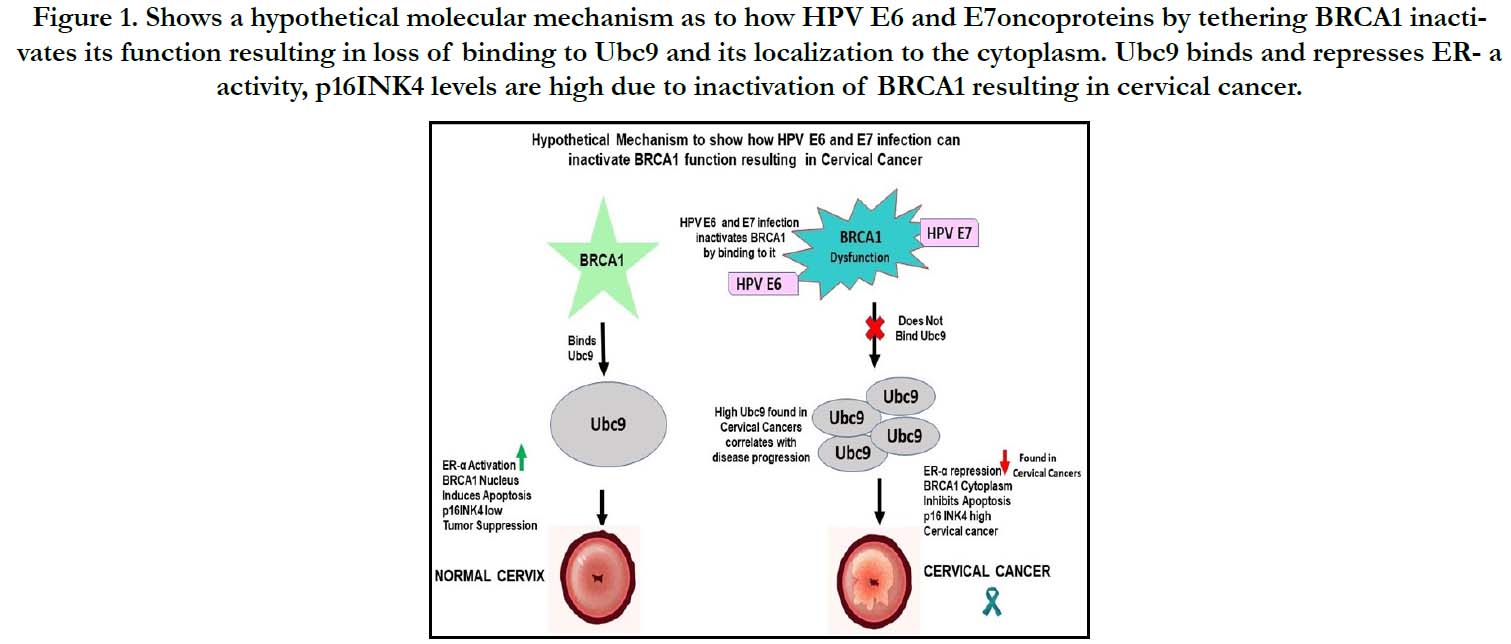

Figure 1. Shows a hypothetical molecular mechanism as to how HPV E6 and E7oncoproteins by tethering BRCA1 inactivates its function resulting in loss of binding to Ubc9 and its localization to the cytoplasm. Ubc9 binds and represses ER- a activity, p16INK4 levels are high due to inactivation of BRCA1 resulting in cervical cancer.
Conclusion
In conclusion several studies have shown an increased risk of
TNBC and cervical cancers in carriers of BRCA1 mutation or
dysfunction caused by HPV infection [2]. Amolecular pathway
as to how BRCA1 mutation/dysfunction results in TNBC was
shown [6, 11] whether a similar mechanism is involved in triggering
cervical cancer following HPV infection needs to be investigated.
Based on this, we hypothesize that HPV oncogenes
interfere with BRCA1 function leading to cervical cancer.The
next step from here is to continue the investigation into the proposed
molecular mechanism involving HPV oncogenes disrupting
BRCA1 functions and study how Ubc9, p16INK and ERa
Can be used as biomarkers for early detection or as potential
therapeutic targets for BRCA1-associated cervical cancers which
can rescue BRCA1 function.
Acknowledgements
This work was supported in part by the Georgia Cancer Coalition
Distinguished Cancer Scholar Award, NIHMD research
endowment award 2S21MD000101, U54 MD007602, and U54
CA118638 to V.N.R. V.N. R�s lab was also supported in part by
funds from the VOYA foundation, EAD Foundation and Breast
cancer partnership grant It�s the Journey Inc, a Cure in our lifetime
and Georgia CORE.
References
-
[1]. American Cancer Society - cancer facts & statistics, n.d. Retrieved August
1, 2022, from https://cancerstatisticscenter.cancer.org/#!/cancer-site/
Cervix?module=g4eIyv7V.
[2]. Mersch J, Jackson MA, Park M, Nebgen D, Peterson SK, Singletary C, et al. Cancers associated with BRCA 1 and BRCA 2 mutations other than breast and ovarian. Cancer. 2015 Jan 15;121(2):269-75.
[3]. Ibeanu OA. Molecular pathogenesis of cervical cancer. Cancer Biol. Ther. 2011 Feb 1;11(3):295-306.
[4]. M�nger K, Scheffner M, Huibregtse JM, Howley PM. Interactions of HPV E6 and E7 oncoproteins with tumour suppressor gene products. Cancer Surv. 1992 Jan 1;12:197-217.
[5]. Yuli C, Shao N, Rao R, Aysola P, Reddy V, Oprea-llies G, et al. BRCA1a has antitumor activity in TN breast, ovarian and prostate cancers. Oncogene. 2007 Sep 6;26(41):6031-7.Pubmed PMID: 17384678.
[6]. Qin Y, Xu J, Aysola K, Begum N, Reddy V, Chai Y, et al. Ubc9 mediates nuclear localization and growth suppression of BRCA1 and BRCA1a proteins. J Cell Physiol. 2011 Dec;226(12):3355-67.Pubmed PMID: 21344391.
[7]. Chen H, Wu J, Zhang Z, Tang Y, Li X, Liu S, et al. Association between BRCA status and triple-negative breast cancer: a meta-analysis. Front. pharmacol. 2018 Aug 21;9:909.
[8]. Zhang Y, Fan S, Meng Q, Ma Y, Katiyar P, Schlegel R, et al. BRCA1 interaction with human papillomavirus oncoproteins. J. Biol. Chem. 2005 Sep 30;280(39):33165-77.
[9]. den Boon JA, Pyeon D, Wang SS, Horswill M, Schiffman M, Sherman M, et al. Molecular transitions from papillomavirus infection to cervical precancer and cancer: Role of stromal estrogen receptor signaling. Proc Natl Acad Sci U S A. 2015 Jun 23;112(25):E3255-64.Pubmed PMID: 26056290. [10]. Yim EK, Park JS. The role of HPV E6 and E7 oncoproteins in HPV-associated cervical carcinogenesis. Cancer Res Treat. 2005 Dec 31;37(6):319-24.
[11]. Xu J, Watkins T, Reddy A, Reddy ES, Rao VN. A novel mechanism whereby BRCA1/1a/1b fine tunes the dynamic complex interplay between SUMOdependent/ independent activities of Ubc9 on E2-induced ERalpha activation/ repression and degradation in breast cancer cells. Int J Oncol. 2009 Apr;34(4):939-49.Pubmed PMID: 19287951.
[12]. Bernier-Villamor V, Sampson DA, Matunis MJ, Lima CD. Structural basis for E2-mediated SUMO conjugation revealed by a complex between ubiquitin-conjugating enzyme Ubc9 and RanGAP1. Cell. 2002 Feb 8;108(3):345-56.Pubmed PMID: 11853669.
[13]. Gostissa M, Hengstermann A, Fogal V, Sandy P, Schwarz SE, Scheffner M, Del Sal G. Activation of p53 by conjugation to the ubiquitin-like protein SUMO-1. EMBO J. 1999 Nov 15;18(22):6462-71.
[14]. Lee JS, Choi HJ, Baek SH. Sumoylation and its contribution to cancer. SUMO regulation of cellular processes. 2017 Feb 15;963:283-98. [15]. Mo YY, Yu Y, Theodosiou E, Rachel Ee PL, Beck WT. A role for Ubc9 in tumorigenesis. Oncogene. 2005 Apr;24(16):2677-83.
[16]. Zhu S, Sachdeva M, Wu F, Lu Z, Mo YY. Ubc9 promotes breast cell invasion and metastasis in a sumoylation-independent manner. Oncogene. 2010 Mar;29(12):1763-72.
[17]. Mattoscio D, Casadio C, Fumagalli M, Sideri M, Chiocca S. The SUMO conjugating enzyme UBC9 as a biomarker for cervical HPV infections. Ecancermedicalscience. 2015 Apr 29;9:534.Pubmed PMID: 26015803. [18]. Finzer P, Aguilar-Lemarroy A, R�sl F. The role of human papillomavirus oncoproteins E6 and E7 in apoptosis. Cancer Lett. 2002 Dec 15;188(1- 2):15-24.
[19]. Yoshida K, Miki Y. Role of BRCA1 and BRCA2 as regulators of DNA repair, transcription, and cell cycle in response to DNA damage. Cancer Sci. 2004 Nov;95(11):866-71.
[20]. Durant ST, Nickoloff JA. Good timing in the cell cycle for precise DNA repair by BRCA1. Cell Cycle. 2005 Sep;4(9):1216-22.Pubmed PMID: 16103751.
[21]. Cao L, Li W, Kim S, Brodie SG, Deng CX. Senescence, aging, and malignant transformation mediated by p53 in mice lacking the Brca1 full-length isoform. Genes Dev. 2003 Jan 15;17(2):201-13.Pubmed PMID: 12533509. [22]. Schuyer M, Berns EM. Is TP53 dysfunction required for BRCA1-associated carcinogenesis? Mol Cell Endocrinol. 1999 Sep 10;155(1-2):143-52.Pubmed PMID: 10580847.
[23]. Khleif SN, DeGregori J, Yee CL, Otterson GA, Kaye FJ, Nevins JR, et al. Inhibition of cyclin D-CDK4/CDK6 activity is associated with an E2Fmediated induction of cyclin kinase inhibitor activity. Proc Natl Acad Sci U S A. 1996 Apr 30;93(9):4350-4.Pubmed PMID: 8633069.
[24]. Klaes R, Friedrich T, Spitkovsky D, Ridder R, Rudy W, Petry U, et al. Overexpression of p16INK4A as a specific marker for dysplastic and neoplastic epithelial cells of the cervix uteri. Int J Cancer. 2001 Apr 15;92(2):276-84.
[25]. Benevolo M, Allia E, Gustinucci D, Rollo F, Bulletti S, Cesarini E, et al. New Technologies for Cervical Cancer Screening 2 (NTCC2) Working Group. Interobserver reproducibility of cytologic p16INK4a /Ki-67 dual immunostaining in human papillomavirus-positive women. Cancer Cytopathol. 2017 Mar;125(3):212-220.Pubmed PMID: 27926800.
[26]. Murphy N, Ring M, Killalea AG, Uhlmann V, O'Donovan M, Mulcahy F, et al. p16INK4A as a marker for cervical dyskaryosis: CIN and cGIN in cervical biopsies and ThinPrep smears. J Clin Pathol. 2003 Jan;56(1):56-63. Pubmed PMID: 12499437.
[27]. Farzanehpour M, Muhammadnejad A, Akhavan S, Emami Razavi AN, Jalilvand S, Salimi V,et al. P16INK4A Immunohistochemistry as a Gold Standard for Cervical Cancer and Precursor Lesions Screening. Iran J Public Health. 2020 Feb;49(2):312-322. PMID: 32461939.
[28]. Lau WM, Ho TH, Hui KM. p16INK4A-silencing augments DNA damage-induced apoptosis in cervical cancer cells. Oncogene. 2007 Sep 6;26(41):6050-60.Pubmed PMID: 17369842.
[29]. Gorrini C, Baniasadi PS, Harris IS, Silvester J, Inoue S, Snow B, et al. BRCA1 interacts with Nrf2 to regulate antioxidant signaling and cell survival. J Exp Med. 2013 Jul 29;210(8):1529-44.
[30]. PG Romagnolo A, F Romagnolo D, I Selmin O. BRCA1 as target for breast cancer prevention and therapy. Anticancer Agents Med Chem (Formerly Current Medicinal Chemistry-Anti-Cancer Agents). 2015 Jan 1;15(1):4-14.
[31]. Popova T, Mani� E, Rieunier G, Caux-Moncoutier V, Tirapo C, Dubois T, et al. Ploidy and large-scale genomic instability consistently identify basallike breast carcinomas with BRCA1/2 inactivation. Cancer Res. 2012 Nov 1;72(21):5454-62.Pubmed PMID: 22933060.
[32]. Scott A, Bai F, Chan HL, Liu S, Slingerland JM, Robbins DJ, et al. p16 loss rescues functional decline of Brca1-deficient mammary stem cells. Cell Cycle. 2017 Apr 18;16(8):759-764.Pubmed PMID: 28278054.
[33]. Wang J, Sampath A, Raychaudhuri P, Bagchi S. Both Rb and E7 are regulated by the ubiquitin proteasome pathway in HPV-containing cervical tumor cells. Oncogene. 2001 Aug 2;20(34):4740-9.Pubmed PMID: 11498796.
[34]. Mattoscio D, Casadio C, Miccolo C, Maffini F, Raimondi A, Tacchetti C, et al. Autophagy regulates UBC9 levels during viral-mediated tumorigenesis. PLoS Pathog. 2017 Mar 2;13(3):e1006262.Pubmed PMID: 28253371.





