The Management of Hereditary Epidermolysis Bullosa: Experience of a Moroccan Sub-Population
Barbach Y*, Baybay H, Chaouche M, Zinoune S, Elloudi S, Mernissi FZ
Dermatology Service, CHU HASSAN II, Fes, Morocco.
*Corresponding Author
Ouiame EL Jouari,
Department of Dermatologym,
University Hospital Hassan II, Fez, Morocco.
Tel: 212645768798
Fax: 212535617687
E-mail: eljouariouiame@gmail.com
Received: June 13, 2018; Accepted: September 24, 2018; Published: September 26, 2018
Citation: El Jouari O, Zaougui AA, Gallouj S, Senhaji G, Farih MH, Mernissi FZ. Isolated Genital Psoriasis. Int J Pediat Health Care Adv. 2018;5(4):84-85. doi: dx.doi.org/10.19070/2572-7354-1800024
Copyright: El Jouari O© 2018. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
Abstract
Hereditary epidermolysis bullosa (EBH) is a heterogeneous group of rare genodermatoses, characterized by cutaneous or mucosal fragility, localized or generalized. These are diseases of varying severity, which can have a significant psychosocial impact. Our Moroccan context is particular by the very frequent consanguinity, and the nutritional and infectious complications sometimes leading to death. Our goal was to describe the peculiarities of EBH in our population epidemiologically, clinically, as well as the search for various complications in order to establish a good therapeutic management.
2.Abbreviations
3.Introduction
4.Materials and Methods
5.Results
6.Discussion
7.Conclusion
8.References
Keywords
Epidermolysis Bullosa; Care; Quality of Life; Complication.
Abbreviations
HEB: Hereditary Epidermolysis Bullosa.
Introduction
Hereditary epidermolysis bullosa (HEB) is rare genodermatose characterized by epithelial fragility leading to the formation of bubbles, cutaneous and mucosal erosions [1]. They would affect about 1/50000 to 1/20000 newborns [2]. The cause of this genodermatosis is a deficiency of one of the proteins involved in anchoring the epidermis to the dermis. The deficient protein determines the level of bubble formation and, therefore, the form and severity of the EB subtype. This is a basis for classification. The resulting form is the result of an autoimmune reaction against collagen VII, the protein anchoring fibrils in the papillary dermis, causing detachment of the epidermis. The clinical severity is variable, ranging from minor functional discomfort to the death of the child, to the culprits responsible for severe disabilities, to the infectious, nutritional, cicatricial and functional complications they cause. The aim of this work is to describe the particularities of HEB in our population epidemiologically, clinically, as well as the search for different complications in order to establish a good therapeutic management.
Materials and Methods
This is a retrospective study conducted on cases of children hospitalized or seen in consultation for hereditary epidermolysis bullosa in the dermatology department of CHU HASSAN II of Fez over a period of 5 years between September 2013 and July 2018.
Results
We identified 8 patients. The average age was 4 years, with extremes of 8 days and 12 years, the average age of onset of symptoms was 3 months, a sex ratio of 1.66. The concept of consanguinity was present in 75% of patients with similar cases in the family in 3 patients. Cutaneous biopsy was performed in half of the patients. The age of onset of the disease was variable: at birth in 6 cases (75%), between 1 month and 1 year in 2 cases (25%). The skin lesions were polymorphous with bubbles and erosions in all patients [Figure 1], milia grains in 2 patients (25%) and atrophic lesions and dystrophic scars in 62.5% of cases. Oral mucosal involvement was present in 5 patients (62.5% of cases) [Figure 2]. Nail dystrophy was noted in 7 cases (87.5% of patients) [Figure 3]. Based on the clinical and pathological data, we were able to classify our patients as dystrophic HEB in 6 cases and as simple HEB in 2 cases. The symptomatic treatment with careful education was recommended in all our patients, the complications found in our patients were anemia in 3 patients, a vitamin and protein deficiency in 3 patients, a staggered delay in a patient and a weight delay in 2 patients, deformity of the hands in 2 patients [Figure 4] and infection of wounds in 5 patients.
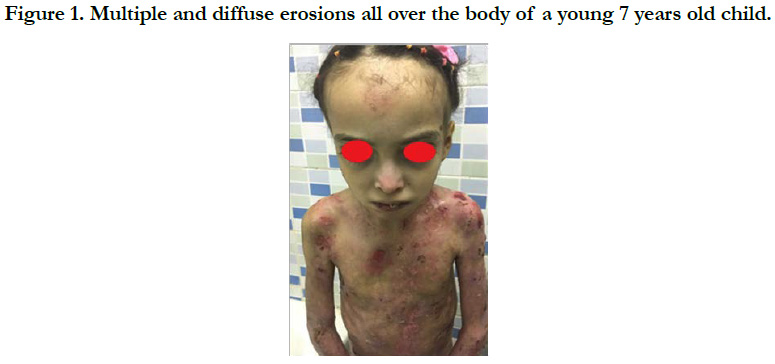
Figure 1. Multiple and diffuse erosions all over the body of a young 7 years old child.
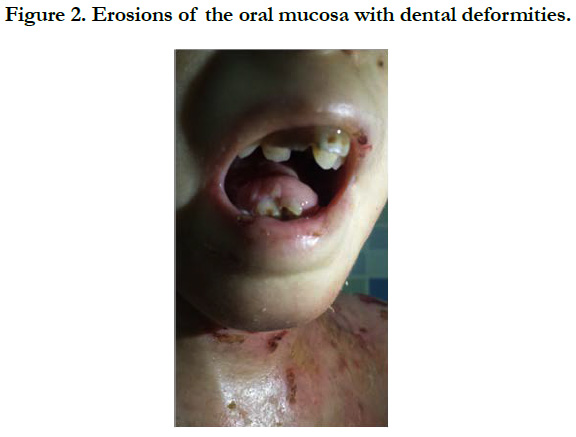
Figure 2. Erosions of the oral mucosa with dental deformities.
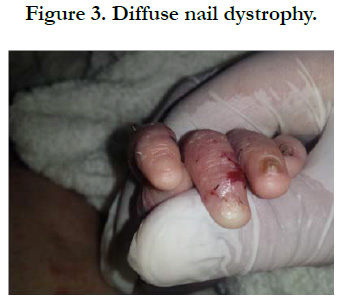
Figure 3. Diffuse nail dystrophy.
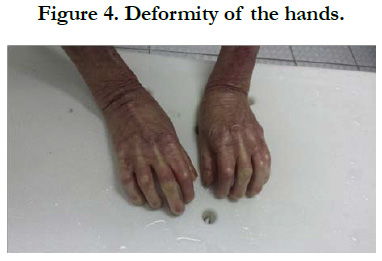
Figure 4. Deformity of the hands.
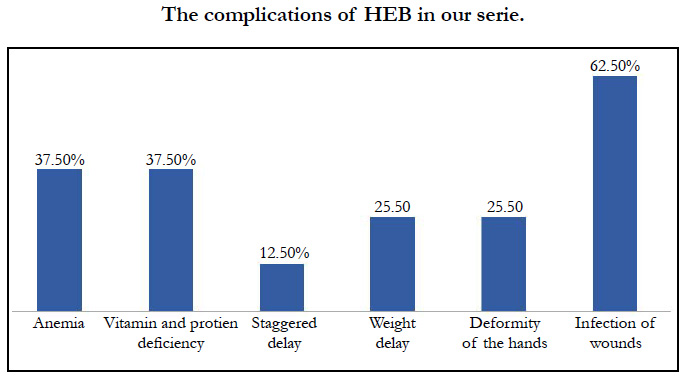
The complications of HEB in our serie.
Discussion
HEB can be classified into three major distinct types, namely, epidermolysis bullosa simplex, junctional epidermolysis bullosa, and dystrophic epidermolysis bullosa, respectively corresponding to a level of cleavage located intraepidermally, within the basement membrane and under the epidermal basement membrane. The latest revision of this classification also identifies Kindler's syndrome as a major subtype, corresponding to a mixed cleavage level [3]. The analysis of the results shows that the clinical lesions presented by our patients corresponded to the characteristic lesions described in the literature [4, 5] Mortality at age 15 is extremely variable, ranging from 0 to 61.8% of deaths according to the type of EBH [4]. The approach of EB patients must be multidisciplinary, at least for the most severe forms. Including not only a dermatologist, a pediatrician nutritionist, nurses trained in wound care, a dietician, a dentist, a physiotherapist, and others. At the same time, it is important to involve parents from the start in caring for their child. Provide them with correct information without being exhaustive. The training of parents in basic gestures necessary for care is the key to better compliance. At the time of diagnosis, the majority of our patients presented complications such as: stun-thyroid retardation found in 2 patients, vitamin deficiencies in 3 patients, infectious complications in 5 patients. This high percentage of complications can be explained in our Moroccan context by the lack of information and awareness of parents causing a delayed diagnosis on one side and the other by the chronicity of the disease and the cost. To cope with this lack of adherence to care, a hospitalization was proposed for this type of patient in the dermatology department involving dermatologists and nurses doctors in order to educate affected children and their parents to improve the quality as well as the life expectancy of these patients. This hospitalization proved to be of a crucial importance in the evolution of these patients, resulting in a good understanding of the relatives of the pathology and thus better adherence to care.
Conclusion
HEB are chronic diseases, often severe, strongly resonating on the quality of life of patients and their families, requiring comprehensive social and medical multidisciplinary management by a team accustomed to these diseases. Staying tuned to the sick and their loved ones is essential to maintain their trust and to maintain a doctor-patient relationship as satisfactory as possible.
References
- Ingen-Housz-Oro S, Blanchet-Bardon C. Hereditary epidermolysis bullosa. EMC-Dermatol, Cosmetol. 2004 Feb 1;1(1):2-18.
- Saurat JH, JPh Lacour, G Meneguzzi. Hereditary epidermolysis bullosa. In: Dermatology and Sexually Transmitted Infections. Masson: 291–296.
- Fine JD, Eady RA, Bauer EA, Bauer JW, Bruckner-Tuderman L, Heagerty A, et al. The classification of inherited epidermolysis bullosa (EB): Report of the Third International Consensus Meeting on Diagnosis and Classification of EB. J Am Acad Dermatol. 2008 Jun;58(6):931-50. doi: 10.1016/j. jaad.2008.02.004. PubMed PMID: 18374450.
- JD Fine, H Hintner. Life with Epidermolysis Bullosa (EB). Etiol, Diagn, Multidiscip Care Ther. New York: Springer; 2009.
- Pai S, Marinkovich MP. Epidermolysis bullosa: new and emerging trends. Am J Clin Dermatol. 2002;3(6):371-80. PubMed PMID: 12113646.





