Immature Sacrococcygeal Teratoma with Microfoci of Yolk Sac Tumor in a Preterm Female Newborn-Is the Surgical Resection Enough?
Roganovic J1*, Ruzman L1, Rimac M1, Jonjic N2
1 Division of Hematology and Oncology, Department of Pediatrics, Clinical Hospital Center Rijeka, Rijeka, Croatia.
2 Department of Pathology, School of Medicine University of Rijeka, Rijeka, Croatia.
*Corresponding Author
Roganovic Jelena, MD, PhD,
Full Professor, Department of Pediatrics, Clinical Hospital Center Rijeka,
School of Medicine, University of Rijeka, Istarska 43, HR-51000 Rijeka, Croatia.
E-mail: roganovic.kbcri@gmail.com
Received: November 06, 2017; Accepted: January 19, 2018; Published: January 24, 2018
Citation: Roganovic J, Ruzman L, Rimac M, Jonjic N. Immature Sacrococcygeal Teratoma with Microfoci of Yolk Sac Tumor in a Preterm Female Newborn- Is the Surgical Resection Enough?. Int J Pediat Health Care Adv. 2018;5(1):70-73. doi: dx.doi.org/10.19070/2572-7354-1800019
Copyright: Roganovic J© 2018. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
Abstract
Sacrococcygeal teratoma (SCT) is the most common neoplasm in neonates and the most common germ cell tumor in children. SCT with malignant elements is very rare in infants. Prenatal diagnosis is of exceptional importance for the adequatefetal intervention, evaluation of perinatal complications and the selection of the optimal treatment. The treatment of choice is complete surgical resection. Postsurgical approach for immature teratoma with malignant elements is not well established. Considering the risk for tumor recurrence, long term follow-up is mandatory. We report clinical, laboratory, radiological and histological findings, surgical procedure, and follow-up in the preterm female neonate with immature SCT with malignant elements.
2.Introduction
3.Case Presentation
4.Discussion
5.Conclusion
6.References
Keywords
Newborn; Teratoma; Yolk Sac Tumor; Sacrococcygeal Region.
Introduction
Germ cell tumors (GCTs) originate from primordial germ cells with multipotent capacity for differentiation. These tumors are very rare in pediatric population, with overall incidence of 0.9 per 100.000 children up to 15 years [1]. GCTs are a heterogeneous group of neoplasms broadly divided in two classes: seminomas, consisting of ovarian dysgerminomas and testicular seminomas, and non-seminomas, which include teratomas, embryonal carcinomas, yolk sac tumors, and choriocarcinomas [2]. GCTs may occur both at gonadal and extragonadal locations. Extragonadal locations are present in about 60% of pediatric cases, with sacrococcygeal region being the most frequent [3, 4]. Approximately 80% of GCTs are benign, while 20% are malignant, representing about 2 to 3% of all malignant pediatric tumors [5]. Two main malignant subtypes of GCTs in children are yolk sac tumor which produces alpha-fetoprotein (AFP) and choriocarcinoma which produces beta-human chorionic gonadotropin (βHCG). Teratomas, the most common GCTs, are comprised of components derived from all three embryonic layers. According to the histology, they are classified as mature, immature and malignant.
We report a case of a premature female newborn with large sacrococcygeal tumor that was first noticed at birth. The child was in a life-threatening condition, requiring urgent diagnostic procedures.
Case Presentation
A female newborn was delivered vaginally at 29th week of gestation due to premature rupture of membranes. She was the second child from the second pregnancy of a healthy 35-yearold woman. Cerclage placement was done at 21th week of gestation because of cervical insufficiency. Fetal ultrasounds were performed regularly and were described as normal. Birth weight, length and head circumference were according to the gestational age (1230 g, 36 cm, and 28 cm respectively). Apgar scores were 6 and 7 at 1 and 5 minutes, respectively. At birth, a sacrococcygeal mass 6 cm in diameter was observed, covered with shiny skin with prominent blood vessels, suspicious for myelomeningocele (Figure 1). Few hours after delivery respiratory distress syndrome developed, that required mechanical ventilation and the surfactant. Because of the risk of perinatal infection, empirical antibiotic therapy with ampicillin and gentamicin was initiated. Intraventricular hemorrhage grade 2 was diagnosed. Considering poor general status of the newborn, urgent diagnostic procedures were done. Plain radiography was performed to exclude congenital spinal abnormalities. Ultrasound demonstrated a large exophytic solid mass with cystic components, calcification, and neovascularization. Ultrasound-guided fine needle aspiration was done; with the cytology finding suggestive for teratoma. AFP level was normal for gestational age (123,420μg/L). Magnetic Resonance Imaging (MRI) of the abdomen and pelvis showed solid-cystic mass with lobulated internal structure, encapsulated towards rectum and delimited towards the uterus and vagina (Figure 2). Distal neural tube defect was excluded, suggesting type 2 SCT according to Altman classification [6]. After cardiopulmonary stabilization, complete tumor resection with coccygectomy was performed on the 18th day of life. Histology demonstrated grade 3 immature teratoma with microfoci of yolk sac tumor (Figure 3). The postoperative course was uneventful. Computed tomography (CT) of the chest and bone scan excluded distant metastases. There were no associated structural urinary or gastrointestinal abnormalities. Control MRI confirmed the absence of residual tumor. The child was discharged at the age of 2 months. She has been followed-up regularly, and at the age of 3 years she is in complete remission with normal urinary function and bowel movements. Because of the cerebral palsy due to intraventricular hemorrhage, the girl is included in the habilitation program.
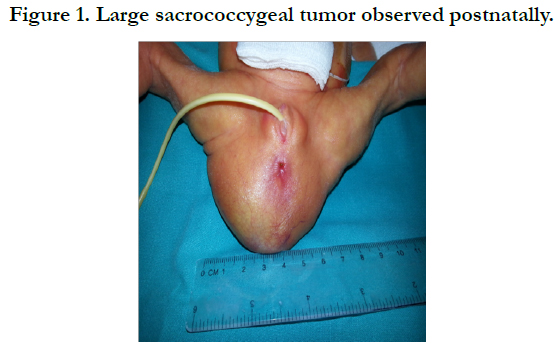
Figure 1. Large sacrococcygeal tumor observed postnatally.
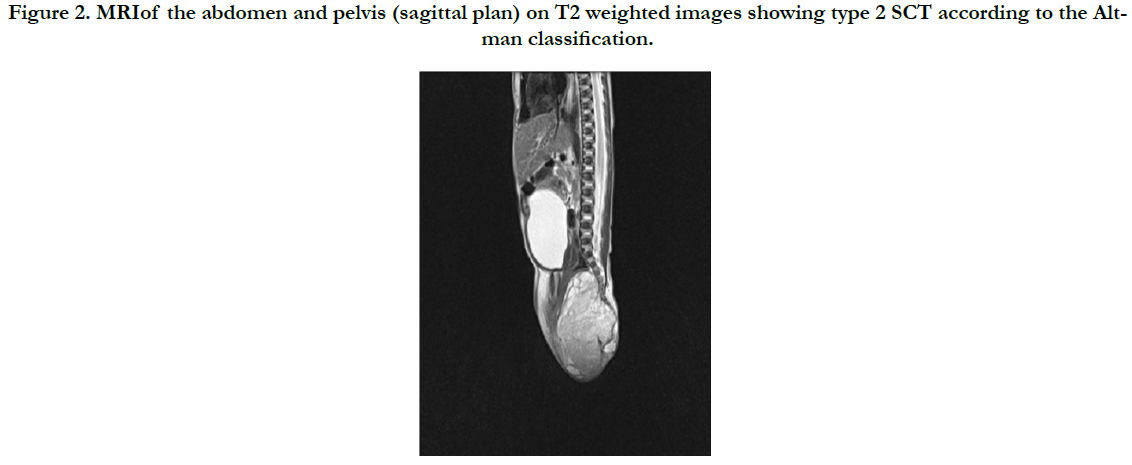
Figure 2. MRIof the abdomen and pelvis (sagittal plan) on T2 weighted images showing type 2 SCT according to the Altman classification.
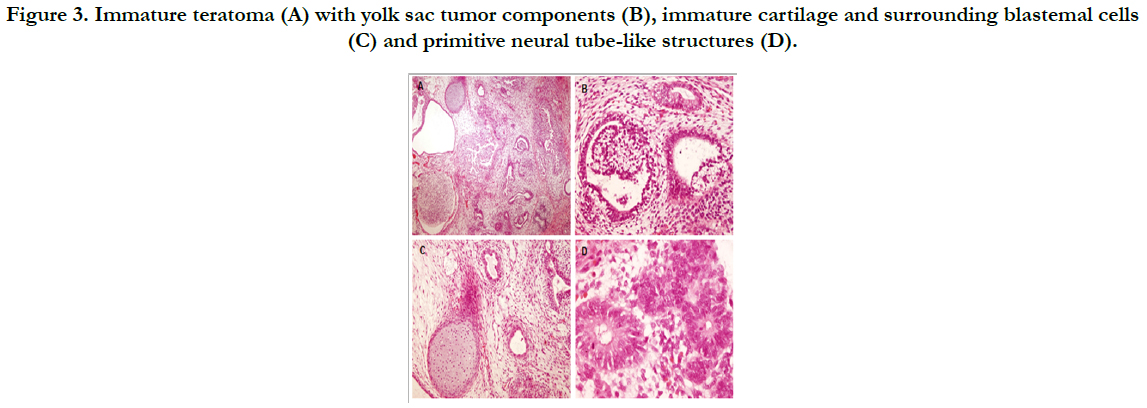
Figure 3. Immature teratoma (A) with yolk sac tumor components (B), immature cartilage and surrounding blastemal cells (C) and primitive neural tube-like structures (D).
Discussion
SCT is a rare tumor, with the prevalence of 1 per 27.000 live births and marked female predisposition [7]. Prenatal diagnosis is established during routine ultrasound in the second trimester in about half of cases [7, 8]. Hydrops fetalis, high-output cardiac failure, and coagulopathy can develop during the fetal life. Perinatal complications include preterm labor, dystocia, spontaneous tumor rupture or hemorrhage, polyhydramnios or oligohydramnios, and maternal mirror syndrome. At birth, sacrococcygeal tumor with or without signs of the rectum and/or bladder obstruction is generally seen. SCT can be associated with the abnormalities of the nervous, cardiac, urogenital or musculoskeletal system. The most important differential diagnosis for perinatal sacrococcygeal mass includes distal neural tube defects (i.e. meningocele, myelomeningocele or myelocystocele) and neurogenic tumors (ganglioneuroma and neuroblastoma) [9]. Lipoma, hamartoma, lymphangioma, hemangioma, enteric cyst, sacral chordoma, and ependymoma are other differential diagnoses of SCT [10].
Etiology of SCT is still not well understood, but is believed to originate from totipotent cells of Hensen’s node and primitive streak. Considering biological capability for continuous embryonic growth, mature teratoma may become malignant. Very few genetic studies have been performed in pediatric teratomas, and no consistent genetic changes have been identified. In contrast, yolk sac tumors of infancy are characterized by gain of chromosomes 1q and 20q, and loss of chromosomes 1p and 6q [11, 12].
Diagnostic imaging of choice is CT or MRI of the abdomen and pelvis, to delineate tumor and to visualize surrounding tissues and organs, especially intraspinal involvement. Chest CT and bone scan is performed for the staging. Detection of AFP may help to confirm the diagnosis. Grading and classification of teratomas is the mainstream for further management. The tumors are classified according to Altman Classification into four types: Type I is predominantly external with a minimal intrapelvic component; Type II is predominantly external with large intrapelvic extension; Type III is predominantly intrapelvic with small external mass, and Type IV is entirely internal. Approximately 45% of teratomas are Type I, 35% are Type II, 10% Type III, and 10% Type IV [6]. WHO classification of GCTs based upon histology distinguishes mature, immature and teratomas with malignant transformation. Immature elements usually contain neuroepithelium, primitive mesenchymal tissue or cartilage. The Gonzales - Crussi grading system [14] refers the grade of immaturity: Grade 0 - mature teratomas with completely differentiated tissue; Grade 1 - teratomas with less than 10% of immature tissue; Grade 2 - teratomas containing among 10% to 50% of immature tissue, and Grade 3 - teratomas consisting of more than 50% of immature tissue. Mature teratomas (G0) are more frequent (54.5%) than immature teratomas (G1-G3, 45.5%). The highest grade of immaturity is present in 7.8% of all teratomas [13].
The cornerstone of the treatment is complete surgical resection [8, 15-18]. Macroscopically, teratomas may be cystic, predominantly solid, or a mixture of solid and cystic components with variable amounts of necrosis [13]. SCTs often do not have a capsule or pseudo capsule, which contributes to the difficulty to achieve complete resection.
The important feature of SCTs is the great rate of relapses. Most relapses occur at the primary site (11% to 49%). Risk factors for recurrence are incomplete resection, immature and malignant histology [16, 19, 20]. Recurrent SCT is usually more undifferentiated/malignant [16]. The 5-year EFS of patients with complete resection is 92%, comparing with 69% in patients with microscopic residues and 38% in patients with macroscopic residual tumor [15]. Therefore, early and complete resection of the primary tumor with coccygectomy must be considered.
SCTs with malignant elements of yolk sac tumor are generally not seen in infants. Only 1.5% of the tumors are malignant when diagnosed and treated in the first week of life, whereas the rate of malignancy is nearly 40% in children older than 1 year [16].
The optimal treatment of patients with completely resected (stage I) immature teratoma with malignant elements is controversial. For many years the standard approach for patients with stage I SCT with malignant elements (yolk sac tumor, embryonal carcinoma or choriocarcinoma) was surgical treatment and chemotherapy [15, 21]. Two therapeutic strategies have been employed: adjuvant chemotherapy after primary resection, or neoadjuvant chemotherapy followed by resection and adjuvant chemotherapy. Most commonly used is carboplatin-etoposidebleomycin regime [17, 22].
Recently, observation without chemotherapy has been suggested. In Pediatric Oncology Group/Children's Cancer Group Study, 4 out of 23 of patients with immature teratoma containing malignant foci who did not receive chemotherapy had recurrence, suggesting that surgical resection followed by close observation is effective treatment [22]. In the study by Gobel et al, 3 patients with stage I malignant SCT did not receive chemotherapy and had no recurrence at 5 years [15]. In recent study by Egler et al., 4 patients with stage I SCT with malignant elements had no recurrence after 3 years of close surveillance [23]. Considering small series of patients in these studies, definite recommendations for treatment of patients with stage I immature teratoma with malignant elements still need to be established.
Long-term follow-up is recommended, including physical examination, tumor marker assessment, and imaging studies. The interpretation of AFP levels should be done in additionto age and clinical parameters, as both benign and immature teratomas may produce modest elevations in AFP and β-HCG [24]. The American Intergroup data showed 5-year OS and EFS for patients with immature teratoma with yolk sac elements of 83% and 80%, respectively [25]. At least one third of patients had some degree of functional impairment, including bowel or bladder incontinence, vesicoureteral reflux, recurrent urinary tract infections, and cosmetic problems [8].
We present a rare case of a newborn with immature SCT containing malignant components. Prenatal diagnosis was missing although ultrasound examinations were performed regularly. Wellplanned complete surgical resection was lifesaving. Considering that no firm guidelines for adjuvant chemotherapy exist, as well as very young age of the patient, and acute and long-term side effects of chemotherapy, we decided to monitor patient closely. At 3-year follow-up, she has no clinical or radiological evidence ofrecurrence, with maintained good urinary and anorectal function.
Conclusion
Newborns with SCT, including rare undifferentiated tumors with malignant elements, have a good prognosis depending on timely recognition and early surgical intervention. Prenatal diagnosis and prenatal close follow-up are of utmost importance, significantly reducing fetal and neonatal morbidity and mortality. Perinatal management requires multidisciplinary team consisting of an obstetrician, neonatologist, radiologist, pediatric surgeon, anesthesiologist, pathologist and pediatric oncologist. Complete surgical excision is the mainstay of the treatment. Optimal treatment of infants with malignant SCT is still evolving. Considering the risk for tumor recurrence, long term follow-up is mandatory.
References
- Cecchetto G. Gonadal germ cell tumors in children and adolescents. Journal of Indian Association of Pediatric Surgeons. 2014 Oct;19(4):189.
- Cushing B, Perlman EJ, Marina NM, Castleberry RP. Principles and Practice of Pediatric Oncology. 5th ed. Pizzo P, Poplack D, editors. Williams and Wilkins(Philadelphia): Germ Cell Tumors; 2006.
- Schneider DT, Terenziani M, Cecchetto G, Olson TA. Gonadal and extragonadal germ cell tumors, sex cord stromal and rare gonadal tumors. InRare tumors in children and adolescents 2012 (pp. 327-402).
- Schneider DT, Brecht IB, Olson TA, Ferrari A, editors. Rare tumors in children and adolescents. Springer Science & Business Media; 2012 Jan 25.
- Rescorla FJ. Pediatric germ cell tumors. InSeminars in pediatric surgery 2012 Feb 29 (Vol. 21, No. 1, pp. 51-60). WB Saunders.
- Altman RP, Randolph JG, Lilly JR. Sacrococcygeal teratoma: American Academy of Pediatrics Surgical Section Survey-1973. J Pediatr Surg. 1974 Jun;9(3):389-98. PubMed PMID:4843993.
- Swamy R, Embleton N, Hale J. Sacrococcygeal teratoma over two decades: birth prevalence, prenatal diagnosis and clinical outcomes. Prenat Diagn. 2008 Nov;28(11):1048-51. doi: 10.1002/pd.2122. PubMed PMID:1897315.
- Gabra HO, Jesudason EC, McDowell HP, Pizer BL, Losty PD. Sacrococcygeal teratoma-a 25-year experience in a UK regional center. J Pediatr Surg. 2006 Sep;41(9):1513-6. PubMed PMID:16952583.
- Yu JA, Sohaey R, Kennedy AM, Selden NR. Terminal myelocystocele and sacrococcygeal teratoma: a comparison of fetal ultrasound presentation and perinatal risk. AJNR Am J Neuroradiol. 2007 Jun-Jul;28(6):1058-60. PubMed PMID:17569957.
- Lemire RJ, Beckwith JB. Pathogenesis of congenital tumors and malformations of the sacrococcygeal region. Birth Defects Research Part A: Clinical and Molecular Teratology. 1982 Apr 1;25(2):201-13.
- Hu J, Schuster AE, Fritsch MK, Schneider DT, Lauer S, Perlman EJ. Deletion mapping of 6q21-26 and frequency of 1p36 deletion in childhood endodermal sinus tumors by microsatellite analysis. Oncogene. 2001 Nov 29;20(55):8042-4. PubMed PMID:11753688.
- Zahn S, Sievers S, Alemazkour K, Orb S, Harms D, Schulz WA, et al., Imbalances of chromosome arm 1p in pediatric and adult germ cell tumors are caused by true allelic loss: a combined comparative genomic hybridization and microsatellite analysis. Genes Chromosomes Cancer. 2006 Nov;45(11):995-1006. PubMed PMID:16897744.
- Harms D, Zahn S, Göbel U, Schneider DT. Pathology and molecular biology of teratomas in childhood and adolescence. Klinische Pädiatrie. 2006 Nov;218(06):296-302.
- Gonzales Crussi F. Extragonadal teratomas. Atlas of tumor pathology. Second series. Washington DC: AFIP. 1982.
- Göbel U, Schneider DT, Calaminus G, Jürgens H, Spaar HJ, Sternschulte W, et al., Multimodal treatment of malignant sacrococcygeal germ cell tumors: a prospective analysis of 66 patients of the German cooperative protocols MAKEI 83/86 and 89. J Clin Oncol. 2001 Apr 1;19(7):1943-50. PubMed PMID: 11283126.
- Derikx JP, De Backer A, van de Schoot L, Aronson DC, de Langen ZJ, van den Hoonaard TL, et al., Factors associated with recurrence and metastasis in sacrococcygeal teratoma. Br J Surg. 2006 Dec;93(12):1543-8. PubMed PMID:17058315.
- Mann JR, Gray ES, Thornton C, Raafat F, Robinson K, Collins GS, et al., Mature and immature extracranial teratomas in children: the UK Children’s Cancer Study Group Experience. J Clin Oncol. 2008 Jul 20;26(21):3590-7. doi: 10.1200/JCO.2008.16.0622. Epub 2008 Jun 9. PubMed PMID:18541896.
- Terenziani M, D'Angelo P, Inserra A, Boldrini R, Bisogno G, Babbo GL, et al., Mature and immature teratoma: A report from the second Italian pediatric study. Pediatr Blood Cancer. 2015 Jul;62(7):1202-8. doi: 10.1002/pbc.25423. Epub 2015 Jan 28. PubMed PMID: 25631333.
- Yao W, Li K, Zheng S, Dong K, Xiao X. Analysis of recurrence risks for sacrococcygeal teratoma in children. Journal of pediatric surgery. 2014 Dec 31;49(12):1839-42.
- Wang Y, Wu Y, Wang L, Yuan X, Jiang M, Li Y. Analysis of Recurrent Sacrococcygeal Teratoma in Children: Clinical Features, Relapse Risks, and Anorectal Functional Sequelae. Med Sci Monit. 2017 Jan 2;23:17-23. PubMed PMID: 28042962.
- Cushing B, Giller R, Cullen JW, Marina NM, Lauer SJ, Olson TA, et al., Randomized comparison of combination chemotherapy with etoposide, bleomycin, and either high-dose or standard-dose cisplatin in children and adolescents with high-risk malignant germ cell tumors: a pediatric intergroup study-Pediatric Oncology Group 9049 and Children's Cancer Group 8882. Journal of Clinical Oncology. 2004 Jul 1;22(13):2691-700.
- Marina NM, Cushing B, Giller R, Cohen L, Lauer SJ, Ablin A, et al., Complete surgical excision is effective treatment for children with immature teratomas with or without malignant elements: A Pediatric Oncology Group/ Children's Cancer Group Intergroup Study. Journal of clinical oncology. 1999 Jul;17(7):2137-.
- Egler RA, Gosiengfiao Y, Russell H, Wickiser JE, Frazier AL. Is surgical resection and observation sufficient for stage I and II sacrococcygeal germ cell tumors? A case series and review. Pediatr Blood Cancer. 2017 May;64(5). doi: 10.1002/pbc.26311. Epub 2016 Oct 27. PubMed PMID: 27786428.
- Barreto MW, Silva LV, Barini R, Oliveira-Filho AG, Sbragia L. Alpha-fetoprotein following neonatal resection of sacrococcygeal teratoma. Pediatr Hematol Oncol. 2006 Jun;23(4):287-91. PubMed PMID:16621769.
- Marina N, London WB, Frazier AL, Lauer S, Rescorla F, Cushing B, et al., Prognostic factors in children with extragonadal malignant germ cell tumors: a pediatric intergroup study. Journal of clinical oncology. 2006 Jun 1;24(16):2544-8.





