A Review of Personalised Molecular Medicine for the Treatment of Corneal Disorders
Courtney DG1*, Thakur A2, Nesbit MA1, Moore JE3, C. B. Tara Moore1
1 School of Biomedical Sciences, University of Ulster, Coleraine, Northern Ireland, UK.
2 British Columbia Cancer Agency, University of British Columbia, Vancouver, BC, Canada.
3 Cathedral Eye Clinic, Belfast, Northern Ireland, UK.
*Corresponding Author
David G. Courtney,
School of Biomedical Sciences,
University of Ulster, Cromore Road,
Coleraine, Northern Ireland, BT521SA, UK.
E-mail: courtney-d@email.ulster.ac.uk
Article Type : Review Article
Received: July 24, 2014; Accepted: August 23, 2015; Published: August 27, 2015
Citation: Courtney DG, Thakur A, Nesbit MA, Moore JE, C. B. Tara Moore (2015) A Review of Personalised Molecular Medicine for the Treatment of Corneal Disorders. Int J Ophthalmol Eye Res. S2:002, 7-18. doi: dx.doi.org/10.19070/2332-290X-SI02002
Copyright: Courtney DG© 2015. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
Abstract
The cornea represents an ideal tissue to elucidate the potential of molecular medicine to treat genetic disorders. It is a small, readily accessible tissue that is easy to monitor treat topically, whilea number of corneal disorders are well characterized with regard to a known genetic cause. In addition, due to their genetic basis most of these conditions are bilateral, affecting both corneas, allowing for a clear comparison between treated and untreated tissue using the fellow eye as a control.
Currently, molecular techniques utilising siRNAs, CRISPR/Cas9, TALENS and ZFNs are being evaluated by many for their therapeutic potential for corneal disorders. Methodologies are discussed within this review with particular emphasis on the more recent and emerging field of CRISPR/Cas9-based therapeutics. Since the advent of gene therapies, effective delivery has been a challenge. Potential ocular delivery techniques include viral vectors, nanoparticles or physical injection.
Studies employing in vitro and in vivo models of corneal disorders have demonstrated promising results for treatment by molecular techniques. However, many of these studies have not translated into the clinic, perhaps due to a lack of good animal models for preclinical testing or failure to develop suitable delivery methods. Until this occurs it is difficult to fully gauge the usefulness of these techniques to treat corneal disorders in human patients over a sustained time period.
Here we present a review of current research into molecular-based therapies for genetic corneal disorders and discuss the applicability of these studies to advancing translational research for other genetic disorders.
2.Introduction
3.Corneal Disorders
4.Gene Therapy
4.1 Germ-line vs Somatic
4.2 siRNAs
4.3 CRISPR/Cas9
4.4 TALENs
4.5 ZFNs
5.Delivery
5.1 Viral vectors
5.2 Direct intrastomal injection
5.3 Nanocarriers
5.4 Electroporation
6.Potential
7.Limitations
8.Conclusions
9.References
Keywords
Cornea; Personalised; Gene; Therapy; CRISPR; Cas9; siRNA; TALEN; ZFN; Delivery.
Introduction
The cornea represents an ideal tissue to be treated using molecular based therapeutics. It is a small transparent a vascular surface, allowing for local non-invasive delivery. The cornea is made up on 3 main layers; starting from the anterior, the epithelium, stroma and endothelium, which are separated by 2 collagenous membranes; Bowman’s layer and Descemet’s membrane. Disorders that affect this tissue also generally result in a loss in visual acuity with clear physical symptoms. This allows for simpler detection of disease resolution in animal models, while if therapies progressed to clinical trials patient visual acuity could easily be assessed as a measure of therapeutic efficaciousness.
As the field of molecular medicine evolves greater numbers of systems are being utilised to ellicit changes in gene expression in an attempt to resolve genetic or environmental disorders. Within this review we focus on 4 of the more common forms of gene therapy utilised to alter gene expression; siRNAs, TALENs, ZFNs and CRISPR/Cas9.
Short-interfering RNAs (siRNAs) are one method of transiently down regulating the expression of any target gene through the exploitation of the RNA interference pathway, often referred to as gene silencing [1]. The bacterial Type II Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)/Cas9 genome editing system is the latest method of interrupting gene expression through cleavage of target DNA. Its effectiveness at cleaving genomic DNA in mammalian cells in vitro and in vivo [2, 3],the specificity that this system exhibits [4, 5] and the relative ease with which targeted systems can be constructed [6], make this method of gene disruption a favourable technique. Transcription activator-like effector nucleases (TALENs) are artificial systems that can be designed and constructed relatively quickly to bind practically any where in the genome and cleave double stranded DNA, thus interrupting the expression of any given target gene [7]. Zinc finger nucleases (ZFNs) constitute a similar artificial system of genome editing. However the construction of these systems is much more time consuming and costly, coupled with the higher probability of off-target cleavage [8]. However, for any of these therapeutic approaches to be a viable alternative to current treatments for corneal disorders, which include invasive corneal keratoplasty, effective delivery systems must be identified and utilised. Due to the location of the cornea, physical delivery systems such as direct injection or topical application are possibilities for the delivery of gene therapies [9], while methods such as electroporation or delivery using viral vectors have previously been shown to be efficacious in delivering DNA constructs to the cornea in vivo [10, 11].
Within this review we discuss the potential corneal disorders that could be treated using molecular based approaches. We also discuss these therapeutic mechanisms in greater detail and their reported use for diseases affecting the ocular surface, while also investigating current delivery methods for these systems. Finally we discuss the potential of molecular medicine to enter the clinic for the treatment of corneal disorders while also elaborating on potential limitations of these systems in their current form.
Corneal Disorders
Corneal disorders encompass a wide range of varying conditions including corneal dystrophies, corneal injury and edema, corneal ectatic diseases. Corneal dystrophies constitute a group of inherited blinding conditions that affect the cornea. Corneal injuries include disorders that arise during the lifetime of a patient due to external factors such as acid/alkali burns, corneal flash burns or scratches on the cornea. Corneal ectatic diseases are disorders, usually arising sporadically in individuals and include keratoconus, age-related macular degeneration and pellucid marginal degeneration.
In the IC3D classification [12] 22 isolated corneal dystrophies were recognised, each with its own genetic causes and clinical outcomes (Table 1). The majority of these conditions are dominantly inherited, therefore individuals suffering from these conditions are heterozygous for the disease causing mutation [12]. These conditions are the most favourable to be treated in a personalised manner by molecular medicine. Techniques aiming to reduce the expression of a disease-causing gene can be utilised to treat these disorders however, only when the gene itself does not display any haploinsufficiency. In other words, when normal cellular function can prevail in the presence of only one functional copy of a gene, the mutated copy of the gene can either be therapeutically silenced or its expression significantly reduced without inducing any detrimental effects.
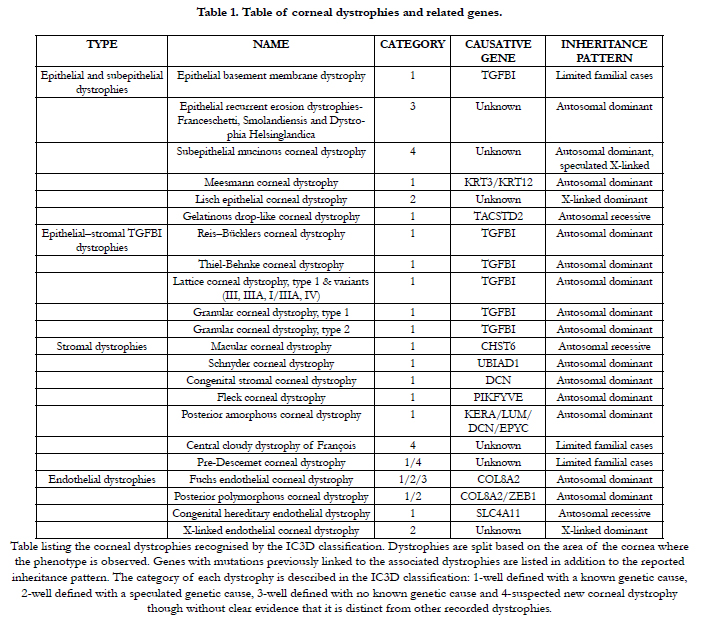

Table 1. Table of corneal dystrophies and related genes.
Table listing the corneal dystrophies recognised by the IC3D classification. Dystrophies are split based on the area of the cornea where the phenotype is observed. Genes with mutations previously linked to the associated dystrophies are listed in addition to the reported inheritance pattern. The category of each dystrophy is described in the IC3D classification: 1-well defined with a known genetic cause, 2-well defined with a speculated genetic cause, 3-well defined with no known genetic cause and 4-suspected new corneal dystrophy though without clear evidence that it is distinct from other recorded dystrophies.
This form of therapy is particularly efficacious for the treatment of dystrophies affecting the corneal epithelium. Due to the speculated high turnover of the human corneal epithelium, from data such as that observed in the rat corneal epithelium (approximately 2 weeks) [13], clearance of mutant disease-causing protein should occur relatively quickly, resulting in a clinically significant change in the patient’s phenotype. Alterations in the expression of disease- causing protein and its clearance may be much slower in the case of corneal stromal and endothelial dystrophies. Especially in the case of Fuchs endothelial corneal dystrophy (FECD), a form of corneal dystrophy resulting in endothelial cell death and the formation of growths, from the Descement membrane, known as guttae [14]. Due to the presence of these seemingly irreversible growths cessation of mutant protein expression would simply halt the progression of the condition, but would not remove the already present aggregates. For this reason, in some cases a corneal graft may first have to be performed before therapies employing molecular based approaches could be effective. A combination of these 2 therapeutic approaches may be ideal for the treatment of Lattice corneal dystrophy Type 1 (LCD1), a condition caused by missense mutations in TGFBI characterised by the formation of amyloid fibrils in the corneal stroma. Current treatments for LCD1 include laser resurfacing keratectomy or surgical keratoplasty [15, 16]. However both these procedures have been shown to result in disease recurrence [15, 16], in some cases within a matter of months, speculatively due to the induction of mutant TGFBI expression in the wound healing process in response to corneal injury incurred during surgery [17, 18].
Corneal injuries and edema are another group of conditions potentially treatable by molecular medicine. Injuries generally result from environmental factors that include corneal burns or infections, while edema can arise from surgery, infection or genetic conditions such as FECD. Corneal injuries result in decreased visual acuity due to the presence of scars or vessels in the otherwise transparent avascular cornea [19]. Some of the main issues surrounding corneal injuries are neovascularisation and the formation of scarring in response to injury. Therapies targeting the expression and activity of vascular endothelial growth factor (VEGF) are currently being investigated for their effectiveness in reducing the onset of neovascularisation due to corneal injury, and the formation of scarring. Biopharmaceuticals including Bevacizumab and Ranibizumab, utilising antibody-based therapies to block the binding of VEGF-A to its receptors, have proven efficacious after subconjunctival injection to the rabbit cornea [20], though side effects have been noted [19]. In addition, a limited number of small molecule studies, targeting downstream pathways induced by VEGF, have demonstrated an ability to reduce corneal neovascularisation in vivo in mice [21]. In comparison corneal edema can result in cloudy vision. If it results from FECD, then a personalised medicine approach treating the underlying genetic cause could prove beneficial in resolving this issue. However, corneal edema resulting from surgery would be more difficult to treat using a molecular approach, as a target gene must be identified. Potential target genes that have previously been associated with corneal edema include N-cadherin [22], TSG-6 [23] and CD147 [24].
Finally corneal ectatic diseases, constitute the fourth form of corneal disorder. Developing molecular-based therapies for corneal ectatic diseases would be much more challenging, due largely to the significant lack of known genetic determinants for these conditions. Keratoconus, the most common corneal ectatic disease, is a progressive non-inflammatory corneal disorder resulting in the distortion of the corneal surface. It presents as a cone like protrusion from the cornea and causes a severe loss in visual acuity [25]. A predisposition to keratoconus has recently been associated with missense mutations in the ZNF469 gene [26, 27], while a number of potential candidate genes, including LOX [28], SOD1 [29], VSX1 [31] and DOCK9 [31], have recently been identified. However, the majority of familial keratoconus cases have no known genetic cause [32-34] making up only 10% of all keratoconus cases, with the remainder seemingly sporadic in nature, apparently arising from environmental factors [12, 32-34]. At present the only available treatments that limit the progression of the disease are hard contact lenses [35], the Intacs corneal inserts or implants [36], and riboflavin/UV-A corneal collagen cross-linking (CXL), first described by Wollensak et al. [37]. CXL is a non-invasive treatment strategy using UV-B light to increase the strength of chemical bonds within the corneal stroma [38], halting the progression of KC for over 6 years [39]. However reservations still persist over the long-term stabilisation of the keratoconic cornea and misgivings exist regarding the use of UV-A, a known DNA mutagen, directly on the cornea [40]. Exposure to UV light has been implicated in the onset of a number of ocular conditions such as pterygium and cataracts, and due to the mutagenic effects of UV-A there is always a risk of corneal tumour formation [41]. If further investigations into the pathomechanism of keratoconus or genetic studies are successful in identifying any viable targets then a molecular medicine based approach to treat keratoconus, or other corneal ectatic diseases, could become a realistic possibility.
Molecular based therapies can be utilised in one of two ways (Figure 1). Genes can be altered in the gametes or fertilised egg (zygote) prior to the development of a blastocyst and implantation. These germ-line modifications greatly increase the probability of the genetic alteration to be present in every cell in the body. This method is used in the production of some transgenic animals, where every cell in the body contains the exact same genetic sequence (Figure 1) [42-44]. Though germ-line editing is much more difficult with greater ethical restrictions [42, 44], a form of germline editing known as 3-person IVF has recently been approved in the UK [45-47]. This technique involves the replacement of mutated maternal mitochondrial DNA, which would lead to mitochondrial disease in the progeny, with mitochondrial DNA from a donor female egg. The nuclear DNA from the gametes remains unaltered, hence the term 3-person IVF [45-47].
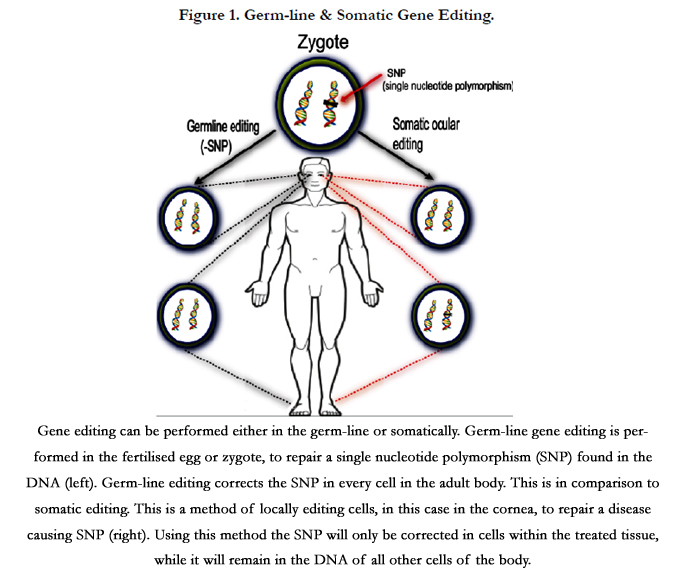
Figure 1. Germ-line & Somatic Gene Editing.
Gene editing can be performed either in the germ-line or somatically. Germ-line gene editing is performed in the fertilised egg or zygote, to repair a single nucleotide polymorphism (SNP) found in the DNA (left). Germ-line editing corrects the SNP in every cell inthe adult body. This is in comparison to somatic editing. This is a method of locally editing cells, in this case in the cornea, to repair a disease causing SNP (right). Using this method the SNP will only be corrected in cells within the treated tissue, while it will remain in the DNA of all other cells of the body.
An alternative method of editing is designed to modify the DNA of cells at a specific site or tissue, in fully formed organisms. This results in a small area of cells containing a different genetic sequence from the majority of cells within the body [48]. This is known as somatic modification (Figure 1). For treating human diseases locally, somatic editing of the genome can be attained through the use of CRISPR/Cas9 [49], TALEN [49, 50] or ZFN [48] based methodologies. Although not having a direct impact on the DNA of cells, siRNAs can be employed locally to alter the normal cellular expression profile.
One method to transiently modify the expression of diseasecausing genes is through the utilisation of siRNAs. These molecules are 22 nucleotide heterodimers introduced into the cellular cytoplasm (Figure 2). In addition, long double-stranded RNA molecules can be delivered to a cell and processed by the endonuclease Dicer, which can cleave these into siRNAs similar to the way endogenously expressed pre-microRNAs are processed [51]. siRNAs can also be created by These siRNA duplexes are taken up by the endogenous RNA-induced silencing complex (RISC), with the strand with the least thermodynamically stable 5’ end (guide strand) being incorporated into this complex, while the remaining strand (passenger strand) is degraded. This process activates the RISC, directing it to cleave single strand mRNA species before they are translated to protein. However, the activated RISC complex can only bind mRNA molecules that are complimentary to the siRNA incorporated into it. The activated RISC complex cleaves the bound mRNA between nucleotides at position 11 and 12 from the 5’ end of the siRNA by the AGO2 endonuclease. This cleaved mRNA molecule is then released from the RISC complex and undergoes further cleavage and degradation by endogenousexonucleases. This RNA interference system is an endogenous method of regulating gene expression through the incorporation of microRNAs. These are endogenously expressed RNA molecules complimentary to specific gene targets, which are used to regulate the expression of these target genes. As stated above, this is a transient method of gene silencing, with each activated RISC complex only having the ability to cleave a limited number of mRNA molecules before undergoing disassociation from the siRNA guide strand (Figure 2).
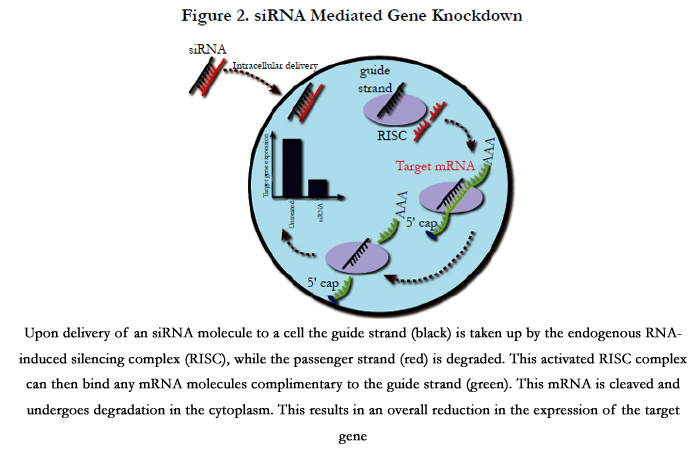
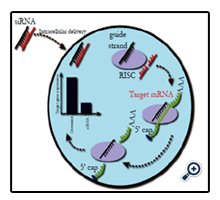
Figure 2. siRNA Mediated Gene Knockdown
Upon delivery of an siRNA molecule to a cell the guide strand (black) is taken up by the endogenous RNA-induced silencing complex (RISC), while the passenger strand (red) is degraded. This activated RISC complex can then bind any mRNA molecules complimentary to the guide strand (green). This mRNA is cleaved and undergoes degradation in the cytoplasm. This results in an overall reduction in the expression of the target gene.
Due to the specificity of this system, where total complementarity of the mRNA to the guide strand of the siRNA is required for cleavage to occur, it has been possible to design siRNAs that distinguish between alleles differing by a single nucleotide and tailor this system to target dominant disease-causing missense mutations. For instance, this allele specificity has also been demonstrated in a mouse model for a keratin related skin disorder [52, 53] and the effectiveness of siRNA therapeutics to treat this condition was further confirmed in a human clinical trial [54]. We, alongside additional researchers, have demonstrated the efficacy of this siRNA-mediated silencing to specifically target and potently reduce the expression of corneal dystrophy causing missense mutation in vitro [55, 56], and have translated 2 of these siRNA molecules into ex vivo disease models [57, 58]. In addition, a number of other studies have demonstrated the effectiveness of siRNA-mediated silencing to treat conditions in the cornea, including Herpes Simplex keratitis [59], corneal scarring [60, 61] and corneal neovascularisation [62, 63].
As previously stated this siRNA-mediated knockdown in gene expression is transient, lasting up to 1 week, and for it to be therapeutically viable the treatment would have to be administered on a regular basis. However, the cornea represents one tissue where this constraint does not restrict the use of this therapeutic system, as topically delivery methods such as nanocarriers, can easily be administered up to several times daily if required. In addition Kim et al., (2005) have shown that delivering a longer dsRNA that requires processing by Dicer can have a sustained silencing effect up to 10 days after initial transfection [64]. These authors also demonstrate that the longer 27 nucleotide complexes do not induce interferon or protein kinase R activity, as has been shown after the introduction of much longer dsRNA [65]. Karpala et al., (2005) have previously discussed the possible antiviral immune responses that may arise due to the introduction of dsRNA into mammalian cells, such as the induction of toll-like receptor 3 pathways and Type 1 interferon [66]. These potential immune responses must always be considered when developing siRNA therapeutics.
Short-hairpin RNAs (shRNAs) are an additional method for altering the expression of a target gene by hijacking the endogenous RISC pathway. shRNAs are expressed from transfected plasmids, and are initially processed by the ribonuclease Drosha, before further processing into an siRNA molecule by Dicer [67]. These siRNA molecules elicit their effects in the same way to synthesised siRNAs, though as the expression construct requires stable integration into the host genome to express some concerns exist over the safety of this system [67].
The transient nature of this system is also beneficial due to its reversibility. If a patient suffers an adverse reaction upon delivery of the siRNA molecules the effect will simply diminish upon degradation of the siRNA. If current research into a more viable delivery system for siRNAs to the cornea is successful [59], this strategy could become a practical option for developing personalised therapeutics for the treatment of corneal disorders.
The CRISPR/Cas9 mechanism of bacterial adaptive immunity is a programmable RNA guided endonuclease, that maintains catalytic activity to induce double strand DNA breaks (DSBs) in mammalian cells [68]. These highly specific and efficient RNAguided DNA endonucleases may be of therapeutic importance for a wide range of diseases [49, 69, 70]. The CRISPR/Cas9 system relies on a single catalytic protein, CRISPR associated protein 9 (Cas9), an endonuclease that is guided to a specific DNA sequence by 2 RNA molecules; the tracrRNA and the crRNA [68]. Since the initial discovery of this system a combined tracrRNA/ crRNA molecule, termed a short guide RNA molecule (sgRNA), has now been established through the utilisation of an artificial loop between the 2 RNA components [71-73]. This has enabled scientists to generate gene editing tools specific for any genomic target much more rapidly than ever before. By altering the 20 nucleotide sgRNA sequence to one complimentary to any particular target, a highly specific DNA editing mechanism can be synthesized in a matter of days [6]. One caveat of this system is that the endonuclease requires a protospacer adjacent motif (PAM), located 3’ of the sgRNA binding site. This PAM sequence is an invariant part of the DNA target but not present in the sgRNA [71, 72]. Its presence directly 3’ of the sgRNA binding site acts as a trigger for Cas9 mediated cleavage of the double-stranded genomic sequence, while its absence results in the inability of Cas9 to cleave the DNA target [74].
For treatment of mutations within the resident corneal stem cells, CRISPR/Cas9 can be utilised in one of two ways (Figure 3). Cleavage of the mutant allele can knockout the expression of the disease-causing gene by non-homologous end joining (NHEJ) and the disease phenotype could resolve, as long as the targeted gene does not display any haploinsufficiency, such as in KRT12 and KRT3 [75]. The other possibility includes the use of a repair oligo completely complimentary to the region surrounding the mutation but including a corrected wild type sequence. This template oligo is then incorporated into the intracellular repair mechanism of homology directed repair (HDR), where the DSB is repaired using this wild type sequence as a template. Although a lower efficiency is achieved using this method, somatic correction of the disease causing mutation and expression of both alleles is sustained [73] (Figure 3). For this system to be a viable therapeutic option for treating corneal dystrophy causing missense mutations, it must differentiate 2 alleles that differ by just a single nucleotide. Recent studies have demonstrated this allele specificity both in vitro in induced pluripotent stem cells [5] and in vivo in transgenic rats [4]. Both studies explored the use of CRISPR/Cas9 to knockout mutant gene function by inducing NHEJ before then studying the induction of HDR when transfections also included single- stranded oligodeoxynucleotides (ssODN), which acted as a repair template.
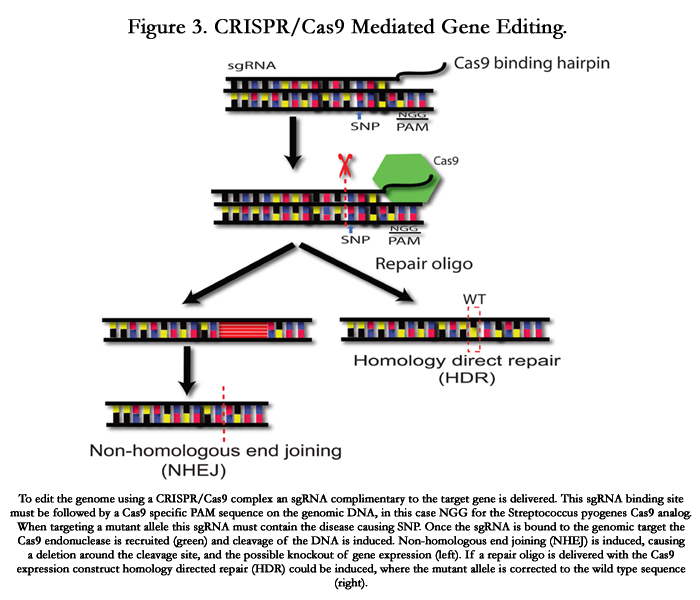
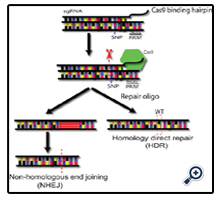
Figure 3. CRISPR/Cas9 Mediated Gene Editing.
To edit the genome using a CRISPR/Cas9 complex an sgRNA complimentary to the target gene is delivered. This sgRNA binding site must be followed by a Cas9 specific PAM sequence on the genomic DNA, in this case NGG for the Streptococcus pyogenes Cas9 analog. When targeting a mutant allele this sgRNA must contain the disease causing SNP. Once the sgRNA is bound to the genomic target the Cas9 endonuclease is recruited (green) and cleavage of the DNA is induced. Non-homologous end joining (NHEJ) is induced, causing a deletion around the cleavage site, and the possible knockout of gene expression (left). If a repair oligo is delivered with the Cas9 expression construct homology directed repair (HDR) could be induced, where the mutant allele is corrected to the wild type sequence (right).
To date two studies have investigated the use of CRISPR/Cas9 to treat ocular disorders in vivo. Courtney et al., (2015) demonstrated the efficacy of an allele specific system utilising a PAM arising due to the presence of a disease causing mutation in KRT12 [76]. This SNP-derived PAM was exploited in vivo in the corneas of mice heterozygous for a Meesmann corneal dystrophy causing KRT12 mutation. This system was found to be both allele specific and potent by both in vitro assays and in vivo assessment. While Wu et al., (2013) investigated the use of an allele specific sgRNA to treat cataracts in a mouse model heterozygous for a 1bp deletion in the Crygc gene. Authors effectively induced germ-line NHEJ and HDR in heterozygous zygotes, where both methods were shown to halt the occurrence of cataracts in adult mice, with this correction confirmed by standard Sanger sequencing [77]. Although somatic alteration of disease causing mutant alleles is more therapeutically relevant to human disorders, the demonstration of the efficacy of this system is very encouraging for future clinical exploits utilising CRISPR/Cas9.
One recently published study by Nihongaki et al., (2015) explored the use of a photoactivatable Cas9 complex, where cleavage could only occur in the presence of blue light [78]. The aim of this study was to increase the safety of Cas9 mediated cleavage by making a locally inducible system, removing the possibility of Cas9 delivered systems inducing cleavage in non-targeted tissues. This system is especially pertinent to specific gene editing of the ocular surface, as blue light can easily be administered locally to the cornea [78]. Short of an efficient delivery system, the research discussed here validates the translation of CRISPR/Cas9 based therapies into trials for the local treatment of the human eye [77].
TALENs are genome editing complexes made up of TALE repeats and a fused FokI endonuclease. Each TALE repeat ismade up of 34 amino acids that recognise a single nucleotidein the target genome [49]. Naturally occurring TALEs were initially utilised to target the genome of plants, which led to the characterisation of this system and the subsequent discovery of TALE binding domains specific to each nucleotide [79, 80]. By fusing the FokI endonuclease to the C-terminal domain of TALE repeats, this novel complex, termed TALEN, can be utilised to cleave DNA in a wide range of in vitro or in vivo models. FokI activity is only achieved by heterodimerization with a second FokI, thus a pair of TALENs is required to achieve cleavage of the target DNA, increasing the specificity of this gene editing system. These TALENs flank the region of interest, a FokI heterodimer is formed and the DNA undergoes cleavage causing aDSB [49, 81]. In a similar manner to CRISPR/Cas9 mediated cleavage, NHEJ is usually induced and results in a genomic insertion or deletion, which can cause a frame shift. Again, in some instances HDR can occur, where a wild type oligo is transfected alongside the TALENs to act as a template for repair.
Although currently no studies employing TALEN based genome editing have been developed for corneal disorders, TALENs have been utilised in vivo to correct a common retinal gene mutation in mice by germ-line editing [82]. In this study fertilised oocytes were injected with mRNA encoding TALENs targeting the homozygous mutant allele of the Crb1 gene along with a repair oligo. Under optimal conditions researchers achieved HDR in 27% of live born mice [82]. This result is encouraging for the development of TALEN based gene therapies, though further optimisation to achieve greater HDR in vivo is required.
ZFNs are another method of permanently editing the DNA of cells. ZFN systems are modular complexes consisting of zinc finger motifs that recognise continuous DNA sequences 3 nucleotides at a time, and a FokI endonuclease to cleave the bound DNA, similar to its utilisation in TALENs [83]. In a similar manner to TALENs, due to the use of FokI, a pair of ZFNs must be delivered to achieve a DSB. However, there are a number of drawbacks to the use of ZFNs in the development of personalised therapeutics. To date there are still a number of nucleotide triplets which no zinc fingers have been discovered to recognise, thus limiting the therapeutic areas in which these complexes can be employed [49]. The process of constructing ZFNs for a novel target is also a slow and costly process [84, 85], especially in the advent of CRISPR/Cas9 genome editing systems that demonstrate comparable editing efficiencies [86].
Two interesting studies investigating the use of ZFNs to treat genetic diseases of the eye endeavoured to design treatments for 2 different mutations that cause retinitis pigmentosa (RP). One study by Greenwald et al., (2010) focused on a common RP causing mutation in the RHO gene [87] while Overlack et al., (2012) concentrated their efforts on a mutation in the USH1C gene that causes Usher syndrome [88]. Both studies demonstrated the effectiveness of ZFNs to bind and cleave mutant alleles in in vitro genetic models of the diseases while also investigating cytotoxic effects, of which there were none. In addition both these studies investigated the induction of HDR to repair the targeted mutation when a wild type template was provided during transfections, which proved efficacious in both cases [87, 88]. Though these initial in vitro findings are promising some issues including the long term safety and effectiveness of these systems still need to be investigated in vivo using animal models of the diseases.
Delivery
Once an established method of achieving a therapeutic response is evaluated in vitro the therapeutic system must be delivered to the cornea and evaluated in vivo. A number of delivery methods have now been utilised to deliver therapeutic molecules to the ocular surface of transgenic animal models including; viral vectors, direct intrastromal injection, nanocarriers and electroporation (Figure 4).
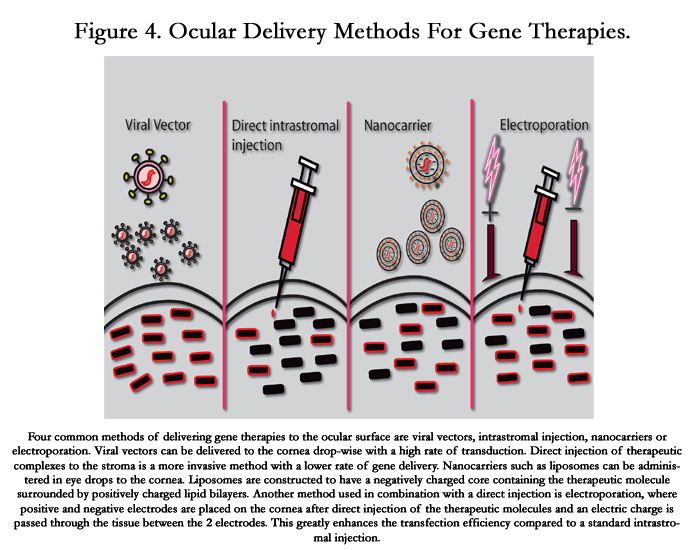
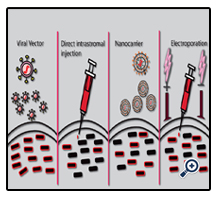
Figure 4. Ocular Delivery Methods For Gene Therapies.
Four common methods of delivering gene therapies to the ocular surface are viral vectors, intrastromal injection, nanocarriers or electroporation. Viral vectors can be delivered to the cornea drop-wise with a high rate of transduction. Direct injection of therapeutic complexes to the stroma is a more invasive method with a lower rate of gene delivery. Nanocarriers such as liposomes can be administered in eye drops to the cornea. Liposomes are constructed to have a negatively charged core containing the therapeutic molecule surrounded by positively charged lipid bilayers. Another method used in combination with a direct injection is electroporation, where positive and negative electrodes are placed on the cornea after direct injection of the therapeutic molecules and an electric charge is passed through the tissue between the 2 electrodes. This greatly enhances the transfection efficiency compared to a standard intrastromal injection.
Viral vectors have readily been used, to great effect, to deliver DNA expression plasmids to cells in vitro. However reservations over the safety of some of these delivery systems have restricted their use in vivo in combination with therapeutic systems.
One major drawback of the use of traditional lentiviral vectors is their innate ability to integrate into the host genome and replicate. One study by Bemelmans et al., (2009) demonstrated, in pig corneas, the efficient transduction of stromal keratocytes after injection of a lentiviral GFP expressing vector, and subsequent culture of the explants [89]. Although sustained GFP expression was observed up to 3 weeks after initial transduction, only the area directly around the injection site successfully expressed the transgene, while no GFP was found in remaining stromal keratocytes. Parker et al., (2007) performed a similar experiment, successfully targeting the corneal endothelium with a GFP expressing lentiviral vector [90]. A study aiming to decrease corneal graft failure in animal models performed by Parker et al., (2010) investigated the therapeutic potential of overexpressing IL10 [91]. Interestingly, this study compared a lentiviral vector with an adenoviral (AV) vector to overexpress IL10 in donor corneas ex vivo. Although this study identified that both vectors could efficiently transduce the cornea, the AV vector was found to induce 1000 fold greater expression of IL10 mRNA in comparison to the lentiviral vector. Of note, the survival rate of lentiviral transduced corneas grafted onto sheep, was extended up to an average of 7 days, and a maximum of 28 days, compared to untreated corneas, demonstrating the potential capabilities of virally delivered gene therapies to the cornea.
Adenoviruses and adeno-associated viral vectors (AAV) are seen as a safer method of deliver, due to their inability to integrate into the host genome [92]. The use of AAVs to deliver gene editing systems to tissues is gaining greater momentum as the efficacy and safety of these vectors is better evaluated (Figure 4). One encouraging aspect of this viral system is the production of a replication deficient AAV, where 2 genomic regions essential for viral replication (E1 and E3) have been deleted [92, 93]. In addition, the infection of humans by AAVs has never led to individuals exhibiting any symptoms or deleterious effects. These safety outcomes have led to the use of this delivery method in a clinical trial for the treatment of the retinal disorder Leber's congenital amaurosis (LCA) [94-96]. This clinical trial delivered a gene, RPE65, to patients with retinal cells lacking expression of functional RPE65. Initially tested in vivo in mouse [97, 98] and canine [99, 100] models of LCA, delivery of this therapeutic system by AAV was found to be effective. Long term follow up of patients 3 years removed from treatment yielded positive results with no safety concerns arising from its administration [101]. In addition to this research, a study by Mohan et al., (2011) investigated the modulation of neovascularisation by the transduction of the decorin gene to rabbit stromal keratocytes using an AAV vector [102]. The authors found that angiogenesis within the cornea was significantly reduced in decorin treated corneas up to 14 days after initial transduction. This research also found that mRNA of pro-angiogenic genes VEGF and MCP-1 was decreased, while the anti-angiogenic gene PEDF was increased, in the decorin delivered corneas when compared to untreated corneas [102]. These studies are encouraging for AAV delivery to the ocular surface and for the utilisation of molecular medicine to correct blinding genetic disorders.
Though AAVs are a more desirable delivery method due to mounting evidence surrounding their safety, adenoviral vectors (AV) have previously demonstrated efficient delivery of a GFP expressing construct to the corneal surface. Jessup et al., (2005) optimised the expression of GFP in corneal endothelial cells, delivered by an adenoviral vector ex vivo, where expression was observed up to 28 days after initial transduction [93]. Interestingly another study aimed at transducing only corneal keratocytes of the stroma utilised a very similar technique [10]. However, in this instance researchers attempted to use a previously identified tissue-specific promoter for the corneal stroma, the keratocanpromotor [103], to drive GFP expression. This study successfully transduced corneal cells, with GFP expression visualised only within the corneal stroma [10]. Saghizadeh et al., (2010) successfully expressed the c-met gene, delivered by an AV vector, ex vivo in human organ-cultured diabetic corneas [104]. The premise of this research was that diabetic corneas display a decrease in the hepatocyte growth factor (HGF) receptor, leading to a decreased wound healing efficiency, and that by increasing c-met expression in the corneas normal HGF signalling could be restored. This interaction of HGF and c-met has previously been shown to be involved in wound healing [105, 106]. Overexpression of AV delivered c-met in the diabetic corneas successfully resolved HGF signalling, while wound healing time was found to have decreased from 6 days to an average of 3 days, closely aligned with the 2.5 days observed in healthy corneas. This research indicates the potential of adenovirus delivered gene therapy constructs to the cornea while also demonstrating the realistic possibility of expressing these therapeutic systems in a tissue specific manner.
Direct injection of therapeutic molecules, such as siRNAs or plasmids, is the simplest method of delivery to the cornea, though is invasive and thus repeat treatments can be problematic (Figure 4). This method of delivering siRNA molecules has been effective in administering and achieving a therapeutic response in a clinical trial for an inherited skin disorder [54]. However due to the transient nature of siRNA mediated knockdown, with a reduction in expression achieved up to 5 days after initial injections in vivo[107], repeated administration is required. This greatly diminishes the potential of delivering siRNAs by direct injection to treat corneal disorders.
However for therapies that alter the expression of genes permanently, thus requiring minimal repeat administration, delivery by direct injection to the corneal stroma is a possibility. Moore et al., (2002) and Stechschulte et al., (2001) have previously demonstrated the transient expression of reporter constructs directly injected into the corneal stroma [9, 108]. Courtney et al., (2015) have also reproduced this in mice using a Cas9-GFP expressing construct injected into the corneal stroma [76]. This method of delivery could be utilised for editing the DNA of resident corneal stem cells through the utilisation of ZFN, TALEN or CRISPR/Cas9 systems. Through the optimisation of this delivery technique the vast majority of corneal stem cells could undergo genomic editing with a minimal number of intrastromal injections. This would significantly increase the potential of applying this technique to administer molecular based gene editing therapies to the cornea.
Drug delivery through the use of nanocarriers is gaining momentum in the current research climate of molecular medicine. In general nanocarriers are metal-based, lipid-based or polymerbased, with each having their own toxicity profiles and physicochemical properties. Either drugs or DNA can easily be incorporated into these complexes, and administration methods including oral, nasal and intra-ocular have all proven effective [86, 87]. Small particles are constructed containing the siRNA molecule or gene editing expression plasmid and could perhaps be applied as an eye drop formulation to the cornea (Figure 4) [111]. This method was recently utilised by Bhela et al (2015), where amiR-155 inhibitor was encased in nanoparticles and delivered in vivo to a mouse cornea infected with herpes simplex virus-1 [112]. This inhibitor reduced vascularisation and the progression of stromal keratitis lesions in the mouse cornea. This study demonstrated the potential efficacy of molecular therapies for treating corneal disorders, delivered by nanoparticles.
One of the main polymers being investigated for its use in nanoparticle formulations is chitosan [113]. The efficacy of chitosanformulated nanoparticles to deliver expression constructs and siRNAs has previously been confirmed in vitro, while toxicity did not appear to be an issue in these studies [111, 114]. Kean et el., (2010) previously discussed the toxicity issues of chitosan/plasmid DNA nanoparticles, stating that toxicity issues can arise due to the charge interaction between DNA and chitosan, affecting cellular uptake and the microenvironment [115]. Chitosan is positively charged, explaining why it has been so effective at harbouring DNA, as it can efficiently form electrostatic interactions with negatively charged nucleic acids [111].
However, lipid-based nanocarriers, or liposomes, appear to be a preferable therapeutic method of nanoparticle delivery to the eye. Liposomes are made up of cationic lipid bilayers surrounding a condensed anionic DNA core. One study by Khurana et al., (2013) found liposomes to be more efficient at delivering siRNA molecules in vitro, when compared to chitosan based nanocarriers [111]. Although there are currently no studies solely investigating the liposome mediated delivery of nucleic acids to the cornea, a significant amount of research has been performed delivering DNA to the retina [116, 117]. However, one study performed by Dai et al., (2013) utilised liposome complexes to deliver an immunosuppressant small molecule drug to rabbit corneas [118]. This study demonstrated the feasibility of liposomes to deliver insoluble drugs to all corneal layers, while concurrently identifying that bile salts, either sodium taurocholate or sodium glycocholate, incorporated into these liposomes greatly increase the distribution of drug release in vivo [118]. This research demonstrated that molecules could be efficiently delivered to the cornea using liposomes, while additional studies have achieved effective delivery of nucleic acids by liposomes to other layers of the eye. These studies appear to identify no immediate adverse effects upon delivery of liposomes to the eye.
One commonly employed method of gene delivery to the cornea is electroporation. Delivery by this method involves an initial injection of the plasmid DNA or siRNA into the cornea. Electrodes are then placed on the cornea and a current is passed between them, similarly to in vitro electroporation (Figure 4). This allows the DNA/siRNA to more readily enter the cells through the electro-permeabilization of the cell membrane [119]. Initial studies aimed to target the corneal endothelium of rats by first administering plasmid DNA into the anterior chamber by direct injection, followed by electroporation of the cornea [120]. This method of gene delivery to the corneal endothelium has since progressed with an optimised method recently being reported for delivery to the mouse corneal endothelium [119]. A further study aimed at achieving gene expression across multiple layers of the cornea was successful in mice [11]. In combination with an intrastromal or subconjuctival injection electroporation was found to increase gene expression 1000 fold, compared to the use of injection alone. This same study also monitored expression of the inflammatory marker interleukin-6 and physically assessed the presence of corneal trauma. No damage to the cornea or inflammation was detected at the optimal field strength of 200V/cm [11].
This method of delivery can be utilised for each of the molecular systems previously stated: siRNAs, CRSIPR/Cas9, TALENs and ZFNs. A study performed by Hao et al., (2009) achieved effective delivery of siRNA molecules to the corneal epithelium of mice [121]. This study applied a solution containing the siRNAs topically to the mouse cornea before electroporation was performed. Although the efficacy of the siRNA to reduce gene expression was not assessed in this study, potent delivery to the corneal epithelium was confirmed by utilisation of a fluorescently labelled siRNA molecule. The physical appearance of the cornea was examined post electroporation with no observable damage detected [121]. Although not delivered to the cornea, the efficacy of CRISPR/Cas9 mediated gene editing after delivery by electroporation has recently been assessed in mouse zygotes [122]. However in this instance the RNA for the CRISPR/Cas9 system was delivered to the cell, in place of a DNA expression construct. This suppresses the ability of the expression construct to integrate and over produce Cas9, which can significantly reduce the potential for off-target effects [122]. The system effectively bound and cleaved the target genomic region after delivery by electroporation. Electroporation appears to be a viable and safe method to deliver molecular systems to the cornea.
Potential
The small surface area of the cornea and the accessibility of the tissue for topical delivery lend itself to the administration of molecular medicine. Gene therapies that ellicit a transient suppression of disease causing genes such as siRNAs, or more permanent alterations in the expression of mutated genes by methods such as CRISPR/Cas9 are currently being developed for ocular disorders [57, 58, 77]. The potential exists for the suppression or correction of any dominantly inherited mutation that leads to the progresThe small surface area of the cornea and the accessibility of the tissue for topical delivery lend itself to the administration of molecular medicine. Gene therapies that ellicit a transient suppression of disease causing genes such as siRNAs, or more permanent alterations in the expression of mutated genes by methods such as CRISPR/Cas9 are currently being developed for ocular disorders [57, 58, 77]. The potential exists for the suppression or correction of any dominantly inherited mutation that leads to the progression of any corneal dystrophy by CRISPR/Cas9 based therapeutics. Previous efforts have demonstrated the potential to deliver expression plasmids to the cornea [93, 108] and these methods could be utilised in the delivery of CRISPR/Cas9 expression constructs to locally edit the DNA of resident corneal stem cells.
These constructs can be tailored to the exact disease causing missense mutation affecting each patient. The design, construction and evaluation of these expression constructs now take a matter of weeks. This form of molecular medicine represents the very real possibility for the establishment of personalised medicine for the treatment of any heterozygous disease-causing mutation saffecting the cornea.
Limitations
As stated previously with respect to keratoconus, one main limitation in the development of molecular based personalised medicines is the need for a predetermined genetic cause. For well characterised conditions such as corneal dystrophies [12] personalised medicines are certainly a viable therapeutic option. However for more complicated conditions, such as corneal burns or corneal ectatic diseases, each individual case is resultant from a variety of environmental or genetic factors. Multifactorial disorders such as these are much more difficult to treat employing a personalised approach as the appropriate therapeutic target can be difficult to deduce.
Aside from this, one limitation that has dominated the field of gene therapy is the development of an effective delivery mechanism for the administration of therapeutic molecules or plasmids. Although, as previously described, a great deal of research has focussed on this issue a large number of these methods are yet to enter clinical trials. However, the nature of the cornea, being a small surface that can be treated topically, removes some of the issues surrounding delivery. Although corneal delivery remains a limitation at present current research is encouraging in the development of gene therapies for corneal disorders.
Finally, a key limiting factor in the development of molecular medicine is the potential for off-target effects. With each of the techniques listed above, siRNAs, TALENs, ZFNs and CRISPR/ Cas9, having previously displayed non specific binding leading to off-target effects. However, as the therapeutic potentials of these techniques are better evaluated off-target effects can be reduced. This is true of CRISPR/Cas9 systems, as described previously, where inducible systems are being developed to try and restrict the directed cleavage of genomic DNA to therapeutically targeted tissues.
Conclusions
Molecular medicine constitutes a promising alternative to current therapies for diseases affecting the cornea. Research is currently focused on developing potential therapies for corneal dystrophies arising from genetic factors and corneal neovascularisation usually resulting from environmental influences. As methods of delivery undergo further investigation, and techniques assessing the specificity of these gene editing approaches become more reliable, the speed at which molecular based therapies can be developedwill increase to the level where personalised medicine may become a reality.
References
- Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, et al. (1998) Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 391(6669): 806-811.
- Kennedy EM, Bassit LC, Mueller H, Kornepati AV, Bogerd HP, et al. (2015) Suppression of hepatitis B virus DNA accumulation in chronically infected cells using a bacterial CRISPR/Cas RNA-guided DNA endonuclease. Virology 476: 196-205.
- Ran FA, Cong L, Yan WX, Scott DA, Gootenberg JS, et al. (2015) In vivo genome editing using Staphylococcus aureus Cas9. Nature 520(7546): 186- 191.
- Yoshimi K, Kaneko T, Voigt B, Mashimo T (2014) Allele-specific genome editing and correction of disease-associated phenotypes in rats using the CRISPR-Cas platform. Nat Commun 5: 4240.
- Smith C, Abalde-Atristain L, He C, Brodsky BR, Braunstein EM, et al. (2015) Efficient and allele-specific genome editing of disease loci in human iPSCs. Mol Ther 23(3): 570-577.
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, et al. (2013) Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8(11): 2281-2308.
- Reyon D, Tsai SQ, Khayter C, Foden JA, Sander JD, et al. (2012) FLASH assembly of TALENs for high-throughput genome editing. Nat Biotechnol 30(5): 460-465.
- Nemudryi AA, Valetdinova KR, Medvedev SP, Zakian SM (2014) TALEN and CRISPR/Cas Genome Editing Systems: Tools of Discovery. Acta Naturae 6(3): 19-40. www.pubmedcentral.nih.gov/articlerender.fcgi?artid =4207558&tool=pmcentrez&rendertype=abstract.
- Stechschulte SU, Joussen AM, von Recum HA, Poulaki V, Moromizato Y, et al. (2001) Rapid ocular angiogenic control via naked DNA delivery to cornea. Invest Ophthalmol Vis Sci 42(9): 1975-1979. www.ncbi.nlm.nih.gov/pubmed/11481260.
- Carlson EC, Liu CY, Yang X, Gregory M, Ksander B, et al. (2004) In vivo gene delivery and visualization of corneal stromal cells using an adenoviral vector and keratocyte-specific promoter. Invest Ophthalmol Vis Sci 45(7): 2194-2200. www.ncbi.nlm.nih.gov/pubmed/15223795 (accessed 8Jul2015).
- Blair-Parks K, Weston BC, Dean DA (2002) High-level gene transfer to the cornea using electroporation. J Gene Med 4(1): 92-100. www.pubmedcentral.nih.gov/articlerender.fcgi?artid=4349340&tool=pmcentrez&rendertype=abstract.
- Weiss JS, Møller HU, Aldave AJ, Seitz B, Bredrup C, et al. (2015) IC3D Classification of Corneal Dystrophies-Edition 2. Cornea 34(2): 117-159.
- Cenedella RJ, Fleschner CR (1990) Kinetics of corneal epithelium turnover in vivo. Studies of lovastatin. Invest Ophthalmol Vis Sci 31(10): 1957-1962.www.ncbi.nlm.nih.gov/pubmed/2210991.
- Eghrari AO, Gottsch JD (2010) Fuchs’ corneal dystrophy. Expert Rev Ophthalmol 5(2): 147-159.
- Ellies P, Renard G, Valleix S, Boelle PY, Dighiero P (2002) Clinical outcome of eight BIGH3-linked corneal dystrophies. Ophthalmology 109(4): 793- 797. www.ncbi.nlm.nih.gov/pubmed/11927442
- Dinh R, Rapuano CJ, Cohen EJ, Laibson PR (1999) Recurrence of corneal dystrophy after excimer laser phototherapeutic keratectomy. Ophthalmology 106(8): 1490-1497.
- Kim TI, Kim T, Kim SW, Kim EK (2008) Comparison of corneal deposits after LASIK and PRK in eyes with granular corneal dystrophy type II. J Refract Surg 24(4): 392-395. www.ncbi.nlm.nih.gov/pubmed/18500090
- Aldave AJ, Sonmez B, Forstot SL, Rayner SA, Yellore VS, et al. (2007) A clinical and histopathologic examination of accelerated TGFBIp deposition after LASIK in combined granular-lattice corneal dystrophy. Am J Ophthalmol 143(3): 416-419.
- Chang JH, Garg NK, Lunde E, Han KY, Jain S, et al. (2012) Corneal neovascularization: an anti-VEGF therapy review. Surv Ophthalmol 57(5):415-429.
- Chen WL, Chen YM, Chu HS, Lin CT, Chow LP, et al. (2014) Mechanisms controlling the effects of bevacizumab (avastin) on the inhibition of early but not late formed corneal neovascularization. PLoS One 9(4): e94205.
- Sun J, Blaskovich MA, Jain RK, Delarue F, Paris D, et al. (2004) Blocking angiogenesis and tumorigenesis with GFA-116, a synthetic molecule that inhibits binding of vascular endothelial growth factor to its receptor. Cancer Res 64(10): 3586-3592.
- Vassilev VS, Mandai M, Yonemura S, Takeichi M (2012) Loss of N-cadherin from the endothelium causes stromal edema and epithelial dysgenesis in the mouse cornea. Invest Ophthalmol Vis Sci 53(11): 7183-7193.
- Kim JA, Ko JH, Ko AY, Lee HJ, Kim MK, et al. (2014) TSG-6 protects corneal endothelium from transcorneal cryoinjury in rabbits. Invest Ophthalmol Vis Sci 55(8): 4905-4912.
- Li S, Nguyen TT, Bonanno JA (2014) CD147 required for corneal endothelial lactate transport. Invest Ophthalmol Vis Sci 55(7): 4673-4681.
- Krachmer JH, Feder RS, Belin MW (1984) Keratoconus and related noninflammatory corneal thinning disorders. Surv Ophthalmol 28(4): 293-322.www.ncbi.nlm.nih.gov/pubmed/6230745.
- Lechner J, Porter LF, Rice A, Vitart V, Armstrong DJ, et al. (2014) Enrichment of pathogenic alleles in the brittle cornea gene, ZNF469, in keratoconus. Hum Mol Genet 23(20): 5527-5535.
- Vincent AL, Jordan CA, Cadzow MJ, Merriman TR, McGhee CN (2014) Mutations in the Zinc Finger Protein Gene, ZNF469, Contribute to the Pathogenesis of Keratoconus. Invest Ophthalmol Vis Sci 55(9): 5629-5635.
- Dudakova L, Liskova P, Trojek T, Palos M, Kalasova S, et al. (2012) Changes in lysyl oxidase (LOX) distribution and its decreased activity in keratoconus corneas. Exp Eye Res 104: 74-81.
- Udar N, Atilano SR, Brown DJ, Holguin B, Small K, et al. (2006) SOD1: a candidate gene for keratoconus. Invest Ophthalmol Vis Sci 47(8): 3345-3351.
- Saee-Rad S, Hashemi H, Miraftab M, Noori-Daloii MR, Chaleshtori MH, et al. (2011) Mutation analysis of VSX1 and SOD1 in Iranian patients with keratoconus. Mol Vis 17: 3128-3136. www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3235532&tool=pmcentrez&rendertype=abstract.
- Burdon KP, Vincent AL (2013) Insights into keratoconus from a genetic perspective. Clin Exp Optom 96(2): 146-154.
- Wang Y, Rabinowitz YS, Rotter JI, Yang H (2000) Genetic epidemiological study of keratoconus: evidence for major gene determination. Am J Med Genet 93(5): 403-409. www.ncbi.nlm.nih.gov/pubmed/10951465.
- Edwards M, McGhee CN, Dean S (2001) The genetics of keratoconus. Clin Experiment Ophthalmol 29(6): 345-351. www.ncbi.nlm.nih.gov/pubmed/11778802.
- Rabinowitz YS (2003) The genetics of keratoconus. Ophthalmol Clin North Am 16(4): 607-620. www.ncbi.nlm.nih.gov/pubmed/14741001.
- Rathi VM, Mandathara PS, Dumpati S (2013) Contact lens in keratoconus. Indian J Ophthalmol 61(8): 410-415.
- Rabinowitz YS (2006) INTACS for keratoconus. Int Ophthalmol Clin 46(3): 91-103. www.pubmedcentral.nih.gov/articlerender.fcgi?artid =3597221&tool=pmcentrez&rendertype=abstract.
- Wollensak G (2006) Crosslinking treatment of progressive keratoconus: new hope. Curr Opin Ophthalmol 17(4): 356-360.
- Vimalin J, Gupta N, Jambulingam M, Padmanabhan P, Madhavan HN (2012) The effect of riboflavin-UV-A treatment on corneal limbal epithelial cells--a study on human cadaver eyes. Cornea 31(9): 1052-1059.
- O’Brart DP, Kwong TQ, Patel P, McDonald RJ, O'Brart NA (2013) Longterm follow-up of riboflavin/ultraviolet A (370 nm) corneal collagen crosslinking to halt the progression of keratoconus. Br J Ophthalmol 97(4):433-437.
- Moore JE, Atkinson SD, Azar DT, Worthington J, Downes CS, et al. (2014) Protection of corneal epithelial stem cells prevents ultraviolet A damage during corneal collagen cross-linking treatment for keratoconus. Br J Ophthalmol 98(2): 270-274.
- Yam JCS, Kwok AK (2014) Ultraviolet light and ocular diseases. Int Ophthalmol 34(2): 383-400.
- Ishii T (2015) Germline genome-editing research and its socioethical implications. Trends Mol Med 21(8): 473-481.
- Baltimore BD, Berg P, Botchan M, Carroll D, Charo RA, et al. (2015) Biotechnology. A prudent path forward for genomic engineering and germline gene modification. Science 348(6230): 36-38.
- Smith KR, Chan S, Harris J (2012) Human germline genetic modification: scientific and bioethical perspectives. Arch Med Res 43(7): 491-513.
- Tavare A (2012) Scientists are to investigate ‘three parent IVF’ for preventing mitochondrial diseases. BMJ 344: e540. www.ncbi.nlm.nih.gov/pubmed/22267657.
- Kouros N (2013) Britain set to be first to allow three-parent IVF. Monash Bioeth Rev 31(2): 24. www.ncbi.nlm.nih.gov/pubmed/24844073.
- Holmes D (2014) New IVF techniques put mitochondrial diseases in focus. Lancet Neurol 13(1): 28-29.
- Rahman SH, Maeder ML, Joung JK, Cathomen T (2011) Zinc-finger nucleases for somatic gene therapy: the next frontier. Hum Gene Ther 22(8):925-933.
- Wei C, Liu J, Yu Z, Zhang B, Gao G, et al. (2013) TALEN or Cas9 - rapid, efficient and specific choices for genome modifications. J Genet Genomics 40(6): 281-289.
- Zhang S, Li L, Kendrick SL, Gerard RD, Zhu H (2014) TALEN-mediated somatic mutagenesis in murine models of cancer. Cancer Res 74(18): 5311- 5321.
- Bartel DP (2004) MicroRNAs : Genomics , Biogenesis , Mechanism , and Function. Cell 116(2): 281-297.
- Hickerson RP, Smith FJ, Reeves RE, Contag CH, Leake D, et al. (2008) Single-nucleotide-specific siRNA targeting in a dominant-negative skin model. J Invest Dermatol 128: 594-605.
- Leachman SA, Hickerson RP, Hull PR, Smith FJ, Milstone LM, et al. (2008) Therapeutic siRNAs for dominant genetic skin disorders including pachyonychia congenita. J Dermatol Sci 51(3): 151-157.
- Leachman SA, Hickerson RP, Schwartz ME, Bullough EE, Hutcherson SL, et al. (2010) First-in-human mutation-targeted siRNA phase Ib trial of an inherited skin disorder. Mol Ther 18(2): 442-446.
- Allen EHA, Atkinson SD, Liao H, Moore JE, Leslie Pedrioli DM, et al. (2013) Allele-Specific siRNA Silencing for the Common Keratin 12 Founder Mutation in Meesmann Epithelial Corneal Dystrophy. Invest Ophthalmol Vis Sci 54(1): 494-502.
- Liao H, Irvine AD, Macewen CJ, Weed KH, Porter L, et al. (2011) Development of allele-specific therapeutic siRNA in Meesmann epithelial corneal dystrophy. PLoS One 6(12): e28582.
- Courtney DG, Atkinson SD, Moore JE, Maurizi E, Serafini C, et al. (2014) Development of Allele-Specific Gene-Silencing siRNAs for TGFBI Arg- 124Cys in Lattice Corneal Dystrophy Type I. Invest Ophthalmol Vis Sci 55(2): 977-985.
- Courtney DG, Atkinson SD, Allen EHA, Moore JE, Walsh CP, et al. (2014) siRNA Silencing of the Mutant Keratin 12 Allele in Corneal Limbal Epithelial Cells Grown From Patients With Meesmann’s Epithelial Corneal Dystrophy. Invest Ophthalmol Vis Sci 55(5): 3352-3360.
- Li Z, Duan F, Lin L, Huang Q, Wu K (2014) A new approach of delivering siRNA to the cornea and its application for inhibiting herpes simplex keratitis. Curr Mol Med 14(9): 1215-1225. http://www.ncbi.nlm.nih.gov/pubmed/25336327.
- Sriram S, Gibson DJ, Robinson P, Pi L, Tuli S, et al. (2014) Assessment of anti-scarring therapies in ex vivo organ cultured rabbit corneas. Exp Eye Res 125: 173-182.
- Sriram S, Robinson P, Pi L, Lewin AS, Schultz G (2013) Triple combination of siRNAs targeting TGFβ1, TGFβR2, and CTGF enhances reduction of collagen I and smooth muscle actin in corneal fibroblasts. Invest Ophthalmol Vis Sci 54(13): 8214-8223.
- Zuo L, Fan Y, Wang F, Gu Q, Xu X (2010) A siRNA targeting vascular endothelial growth factor-A inhibiting experimental corneal neovascularization. Curr Eye Res 35(5): 375-384.
- Kim B, Tang Q, Biswas PS, Xu J, Schiffelers RM, et al. (2004) Inhibition of ocular angiogenesis by siRNA targeting vascular endothelial growth factor pathway genes: therapeutic strategy for herpetic stromal keratitis. Am J Pathol 165(6): 2177-2185.
- Kim DH, Behlke MA, Rose SD, Chang MS, Choi S, et al. (2005) Synthetic dsRNA Dicer substrates enhance RNAi potency and efficacy. Nat Biotechnol 23(2): 222-226.
- DeWitte-Orr SJ, Mehta DR, Collins SE, Suthar MS, Gale M Jr, et al. (2009) Long double-stranded RNA induces an antiviral response independent of IFN regulatory factor 3, IFN-beta promoter stimulator 1, and IFN. J Immunol 183(10): 6545-6553.
- Karpala AJ, Doran TJ, Bean AG (2005) Immune responses to dsRNA: implications for gene silencing technologies. Immunol Cell Biol 83(3): 211- 216.
- Wang Z, Rao DD, Senzer N, Nemunaitis J (2011) RNA interference and cancer therapy. Pharm Res 28(12): 2983-2995.
- Hsu PD, Lander ES, Zhang F (2014) Development and Applications of CRISPR-Cas9 for Genome Engineering. Cell 157(6): 1262-1278.
- Ma Y, Zhang L, Huang X (2014) Genome modification by CRISPR/Cas9. FEBS J 281(23): 5186-5193.
- Zhang F, Wen Y, Guo X (2014) CRISPR/Cas9 for genome editing: progress, implications and challenges. Hum Mol Genet 23: R40-46.
- Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, et al. (2014) Genome- scale CRISPR-Cas9 knockout screening in human cells. Science 343(6166): 84-87.
- Wang T, Wei JJ, Sabatini DM, Lander ES (2014) Genetic screens in human cells using the CRISPR-Cas9 system. Science 343(6166): 80-84.
- Cong L, Ran FA, Cox D, Lin S, Barretto R, et al. (2013) Multiplex genome engineering using CRISPR/Cas systems. Science 339(6121): 819-823.
- Westra ER, Semenova E, Datsenko KA, Jackson RN, Wiedenheft B, et al. (2013) Type I-E CRISPR-cas systems discriminate target from non-target DNA through base pairing-independent PAM recognition. PLoS Genet 9(9): e1003742.
- Kao WW, Liu CY, Converse RL, Shiraishi A, Kao CW, et al. (1996) Keratin 12-deficient mice have fragile corneal epithelia. Invest Ophthalmol Vis Sci 37(13): 2572-2584. www.ncbi.nlm.nih.gov/pubmed/8977471.
- Courtney DG, Moore JE, Atkinson SD, Maurizi E, Allen EH et al. (2015) CRISPR/Cas9 DNA cleavage at SNP-derived PAM enables both in vitro and in vivo KRT12 mutation-specific targeting. Gene Ther doi:10.1038/gt.2015.82
- Wu Y, Liang D, Wang Y, Bai M, Tang W, et al. (2013) Correction of a genetic disease in mouse via use of CRISPR-Cas9. Cell Stem Cell 13(6):659-662.
- Nihongaki Y, Kawano F, Nakajima T, Sato M (2015) Photoactivatable CRISPR-Cas9 for optogenetic genome editing. Nat Biotechnol 33(7):755-760.
- Kay S, Hahn S, Marois E, Hause G, Bonas U (2007) A bacterial effector acts as a plant transcription factor and induces a cell size regulator. Science 318(5850): 648-651.
- Römer P, Hahn S, Jordan T, Strauss T, Bonas U, et al. (2007) Plant pathogen recognition mediated by promoter activation of the pepper Bs3 resistance gene. Science 318(5850): 645-648.
- Campbell JM, Hartjes KA, Nelson TJ, Xu X, Ekker SC (2013) New and TALENted genome engineering toolbox. Circ Res 113(6): 571-587.
- Low BE, Krebs MP, Joung JK, Tsai SQ, Nishina PM, et al. (2014) Correction of the Crb1rd8 allele and retinal phenotype in C57BL/6N mice via TALEN-mediated homology-directed repair. Invest Ophthalmol Vis Sci 55(1): 387-395.
- Kim YG, Cha J, Chandrasegaran S (1996) Hybrid restriction enzymes: zinc finger fusions to Fok I cleavage domain. Proc Natl Acad Sci U S A 93(3): 1156-1160. www.pubmedcentral.nih.gov/articlerender.fcgi?artid=40048&tool=pmcentrez&rendertype=abstract.
- Cermak T, Doyle EL, Christian M, Wang L, Zhang Y, et al. (2011) Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res 39(12): e82.
- Urnov FD, Rebar EJ, Holmes MC, Zhang HS, Gregory PD (2010) Genome editing with engineered zinc finger nucleases. Nat Rev Genet 11(9):636-646.
- Hwang WY, Fu Y, Reyon D, Maeder ML, Tsai SQ, et al. (2013) Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat Biotechnol 31(3): 227-229.
- Greenwald DL, Cashman SM, Kumar-Singh R (2010) Engineered zinc finger nuclease-mediated homologous recombination of the human rhodopsin gene. Invest Ophthalmol Vis Sci 51(12): 6374-6380.
- Overlack N, Goldmann T, Wolfrum U, Nagel-Wolfrum K (2012) Gene repair of an Usher syndrome causing mutation by zinc-finger nuclease mediated homologous recombination. Invest Ophthalmol Vis Sci 53(7): 4140-4146.
- Bemelmans AP, Arsenijevic Y, Majo F (2009) Efficient lentiviral gene transfer into corneal stroma cells using a femtosecond laser. Gene Ther 16(7): 933-938.
- Parker DG, Kaufmann C, Brereton HM, Anson DS, Francis-Staite L, et al. (2007) Lentivirus-mediated gene transfer to the rat, ovine and human cornea. Gene Ther 14(9): 760-767.
- Parker DG, Coster DJ, Brereton HM, Hart PH, Koldej R, et al. (2010) Lentivirus-mediated gene transfer of interleukin 10 to the ovine and human cornea. Clin Experiment Ophthalmol 38(4): 405-413.
- Kotterman MA, Schaffer DV (2014) Engineering adeno-associated viruses for clinical gene therapy. Nat Rev Genet 15(7): 445-451.
- Jessup CF (2005) In vitro adenovirus mediated gene transfer to the human cornea. Br J Ophthalmol 89(6): 658-661.
- Hauswirth WW, Aleman TS, Kaushal S, Cideciyan AV, Schwartz SB, et al. (2008) Treatment of leber congenital amaurosis due to RPE65 mutations by ocular subretinal injection of adeno-associated virus gene vector: short-term results of a phase I trial. Hum Gene Ther 19(10): 979-990.
- Bainbridge JW, Smith AJ, Barker SS, Robbie S, Henderson R, et al. (2008) Effect of gene therapy on visual function in Leber’s congenital amaurosis. N Engl J Med 358(21): 2231-2239.
- Maguire AM, Simonelli F, Pierce EA, Pugh EN Jr, Mingozzi F, et al. (2008) Safety and efficacy of gene transfer for Leber’s congenital amaurosis. N Engl J Med 358(21): 2240-2248.
- Pang J, Chang B, Kumar A, Nusinowitz S, Noorwez SM, et al. (2006) Gene therapy restores vision-dependent behavior as well as retinal structure and function in a mouse model of RPE65 Leber congenital amaurosis. Mol Ther 13(3): 565-572.
- Lai CM, Yu MJ, Brankov M, Barnett NL, Zhou X, et al. (2004) Recombinant adeno-associated virus type 2-mediated gene delivery into the Rpe65 -/- knockout mouse eye results in limited rescue. Genet Vaccines Ther 2(1):3.
- Aguirre GK, Komáromy AM, Cideciyan AV, Brainard DH, Aleman TS, et al. (2007) Canine and human visual cortex intact and responsive despite early retinal blindness from RPE65 mutation. PLoS Med 4(6): e230.
- Acland GM, Aguirre GD, Ray J, Zhang Q, Aleman TS, et al. (2001) Gene therapy restores vision in a canine model of childhood blindness. Nat Genet 28(1): 92-95.
- Jacobson SG, Cideciyan AV, Ratnakaram R, Heon E, Schwartz SB, et al. (2012) Gene therapy for leber congenital amaurosis caused by RPE65 mutations: safety and efficacy in 15 children and adults followed up to 3 years. Arch Ophthalmol 130(1): 9-24. doi:10.1001/archophthalmol.2011.298
- Mohan RR, Tovey JC, Sharma A, Schultz GS, Cowden JW, et al. (2011) Targeted decorin gene therapy delivered with adeno-associated virus effectively retards corneal neovascularization in vivo. PLoS One 6(10): e26432.
- Liu C, Arar H, Kao C, Kao WW (2000) Identification of a 3.2 kb 5’-flanking region of the murine keratocan gene that directs beta-galactosidase expression in the adult corneal stroma of transgenic mice. Gene 250(1-2):85-96. www.ncbi.nlm.nih.gov/pubmed/10854782.
- Saghizadeh M, Kramerov AA, Yu FS, Castro MG, Ljubimov AV (2010) Normalization of wound healing and diabetic markers in organ cultured human diabetic corneas by adenoviral delivery of c-Met gene. Invest Ophthalmol Vis Sci 51(4): 1970-1980.
- Neuss S, Becher E, Wöltje M, Tietze L, Jahnen-Dechent W (2004) Functional expression of HGF and HGF receptor/c-met in adult human mesenchymal stem cells suggests a role in cell mobilization, tissue repair, and wound healing. Stem Cells 22(3): 405-414.
- Chmielowiec J, Borowiak M, Morkel M, et al. (2007) c-Met is essential for wound healing in the skin. J Cell Biol 177(1): 151-162.
- Wang Q, Ilves H, Chu P, Contag CH, Leake D, et al. (2007) Delivery and inhibition of reporter genes by small interfering RNAs in a mouse skin model. J Invest Dermatol 127(11): 2577-2584.
- Moore JE, McMullen TCB, Campbell IL, Richard Rohan, Yuichi Kaji, et al. (2002) The Inflammatory Milieu Associated with Conjunctivalized Cornea and Its Alteration with IL-1 RA Gene Therapy. Invest Ophthalmol Vis Sci 43: 2905-2915. iovs.arvojournals.org/article.aspx?articleid=2162624.
- Wang Y, Rajala A, Rajala RVS (2015) Lipid Nanoparticles for Ocular Gene Delivery. J Funct Biomater 6(2): 379-394.
- Farjo R, Skaggs J, Quiambao AB, Cooper MJ, Naash MI (2006) Efficient non-viral ocular gene transfer with compacted DNA nanoparticles. PLoS One 1: e38.
- Khurana B, Goyal AK, Budhiraja A, Aora D, Vyas SP (2013) Lipoplexes versus nanoparticles: pDNA/siRNA delivery. Drug Deliv 20(2): 57-64.
- Bhela S, Mulik S, Gimenez F, Reddy PB, Richardson RL, et al. (2015) Role of miR-155 in the pathogenesis of herpetic stromal keratitis. Am J Pathol 185(4): 1073-1084.
- Hao J, Li SK, Kao WWY, Liu CY (2010) Gene delivery to cornea. Brain Res Bull 81(2-3): 256-261.
- Howard KA, Rahbek UL, Liu X, Damgaard CK, Glud SZ, et al. (2006) RNA interference in vitro and in vivo using a novel chitosan/siRNA nanoparticle system. Mol Ther 14(4): 476-484.
- Kean T, Thanou M (2010) Biodegradation, biodistribution and toxicity of chitosan. Adv Drug Deliv Rev 62(1): 3-11.
- Rajala A, Wang Y, Zhu Y, Ranjo-Bishop M, Ma JX, et al. (2014) Nanoparticle- assisted targeted delivery of eye-specific genes to eyes significantly improves the vision of blind mice in vivo. Nano Lett 14(9): 5257-5263.
- Adijanto J, Naash MI (2015) Nanoparticle-based technologies for retinal gene therapy. Eur J Pharm Biopharm doi:10.1016/j.ejpb.2014.12.028.
- Dai Y, Zhou R, Liu L, Lu Y, Qi J, et al. (2013) Liposomes containing bile salts as novel ocular delivery systems for tacrolimus (FK506): in vitro characterization and improved corneal permeation. Int J Nanomedicine 8:1921-1933.
- Nickerson JM, Getz SE, Sellers JT, Chrenek MA, Goodman P, et al. (2014) DNA delivery in adult mouse eyes: an update with corneal outcomes. MethodsMol Biol 1121: 165-177.
- Oshima Y, Sakamoto T, Yamanaka I, Nishi T, Ishibashi T, et al. (1998) Targeted gene transfer to corneal endothelium in vivo by electric pulse. Gene Ther 5(10): 1347-1354.
- Hao J, Li SK, Liu CY, Kao WW (2009) Electrically assisted delivery of macromolecules into the corneal epithelium. Exp Eye Res 89(6): 934-941.
- Hashimoto M, Takemoto T (2015) Electroporation enables the efficient mRNA delivery into the mouse zygotes and facilitates CRISPR/Cas9-based genome editing. Sci Rep 5: 11315.





