Vision Loss Case of Bardet Biedl Syndrome
Alzahrani M1*, Rehman MA2, Bahttab S3
1 Resident, Department of Ophthalmology, East Jeddah Hospital, Jeddah, Saudi Arabia.
2 Senior Specialist, Department of Ophthalmology, East Jeddah Hospital, Jeddah, Saudi Arabia.
3 Ophthalmology consultant, Department of Ophthalmology, East Jeddah Hospital, Jeddah, Saudi Arabia.
*Corresponding Author
Mazen Alzahrani,
Resident, Department of Ophthalmology,
East Jeddah Hospital, Jeddah, Saudi Arabia.
E-mail: D.mazen@hotmail.com
Received: January 22, 2018; Accepted: February 28, 2018; Published: March 01, 2018
Citation: Alzahrani M, Rehman MA, Bahttab S. Vision Loss Case of Bardet Biedl Syndrome. Int J Oph thalmol Eye Res. 2018;6(3):374-376. doi: dx.doi.org/10.19070/2332-290X-1800075
Copyright: Alzahrani M© 2018. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
Abstract
We would like to report a rare case of Bardet Biedl Syndrome of a women forty-three years, known case of Retinitis pigmentosa presented for medical report, after ocular and systemic assessment we found she is obese, moon face, and has extra toes of both feet and some other finding which was diagnosed later on Bardet Biedl Syndrome. We documented this rare case for its important in clinical practice for regular systemic follow up and therefore, subsequently requiring management accordingly.
2.Introduction
3.Case Report
3.1 Ocular History
4.Discussion
5.Conclusion
6.References
Keywords
Bardet Biedl; Retinitis Pigmentosa; Obesity; Polydactyly.
Introduction
Bardet Biedl Syndrome is a rare hereditary autosomal recessive disorder with multisystem involvement. The prevalence 1:140,000 and 1:160,000 [1, 2]. While the number prevalence increased with consanguinity marriage by 1:65,000 [3]. The complete BBS represented by obesity, retinal rods and cons degeneration, mental retardation, postaxial polydactyly, stunted growth, renal involvement and hypogonadism [4]. The review would give the fundamental familiarity with the syndrome manifestation that will have an effect in early control of disease and directing advantages to the patients and their families. Hence, we conducted this study along with clinical profile of Bardet Biedl Syndrome patient.
Case Report
A women of forty-three years, known case of retinitis pigmentosa and presented to East Jeddah Hospital, Ophthalmology department requesting a medical report, by history, she was not able to see at night for years then both day and night lost with noticed horizontal nystagmus. Patient has past history learning difficulties. The rest of systemic review is unremarkable. By examination: patient underwent for dysmorphology examination, anthropometric measurements, ophthalmology evaluation such microscopic examination, fundus camera and Optical Coherence Tomography (OCT). Patient was conscious, mild cognitive impairment and no cerebellar signs, including coordination and gait instability. Examinations of power, tone grades and Sensation of all limbs were intact.
Her BMI is 30.8 obese, other findings were polydactyly of lower limbs, Figure 1 upper limb polydactyly has had excised at childhood. The rest of examination was irrelevant. Laboratory and radiology have obtained, include of complete blood count, random blood sugar, HgA1c, liver, renal function tests, and abdominal/ pelvic ultrasonography, Electrocardiography (ECG) that were found to be normal.
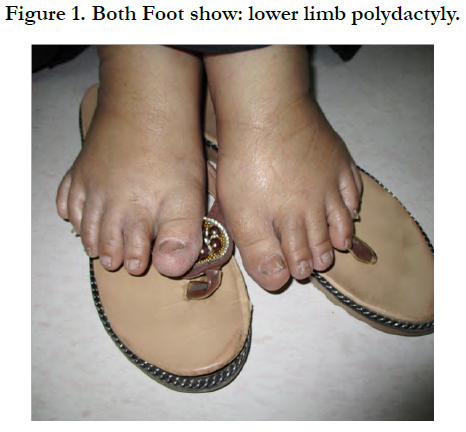
Figure 1. Both Foot show: lower limb polydactyly.
Visual Acuity: light perception both eyes and horizontal nystagmus in both eyes.
Slit Lamp: Lids/Lashes, Conjunctiva and Cornea are Normal.
Goldman Tonometry: OD: 16 mmHg OS: 14 mmHg at 2:30 PM.
Fundus Exam: multiple bony spicules and attenuated vessels with pale optic disc.
Retinitis pigmentosa on fundoscopy Figure 2 thinned and atrophy of RBE Figure 3.
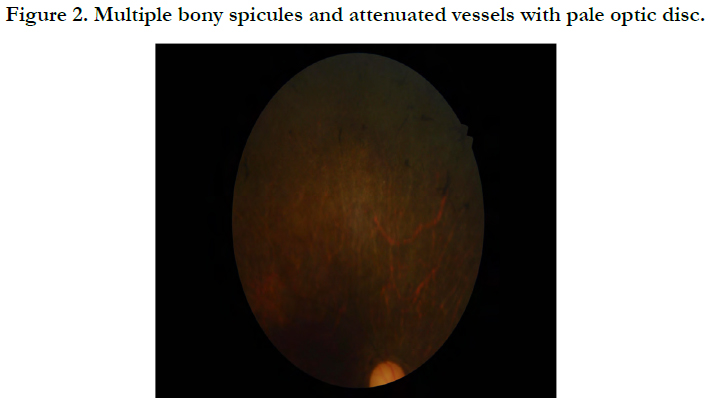
Figure 2. Multiple bony spicules and attenuated vessels with pale optic disc.
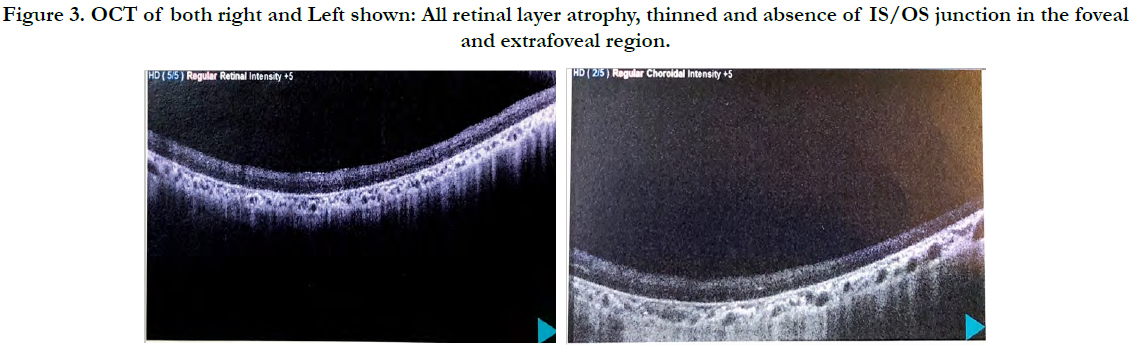

Figure 3. OCT of both right and Left shown: All retinal layer atrophy, thinned and absence of IS/OS junction in the foveal and extrafoveal region.
Discussion
Bardet Biedl Syndrome is a rare hereditary autosomal recessive disorder with multisystem involvement. Diagnosis of BBS was made when four of the primary features or three primary features and two secondary features were present [5]. The primary features of BBS include rod-cone dystrophy, post axial polydactyly or other dystrophic extremities such brachydactyly or syndactyly, obesity, impaired cognitive, renal abnormality, and male hypogonadism [6]. The other signs associated secondary features include liver fibrosis, diabetes mellitus, dyslipidemia, short stature, speech defects, and developmental delay.
In contrast, to the variety of manifestations among BBS, the rodcone deterioration prevalence still the highest number reached up to 95% our patient was started complain in early second decade of age for only night blindness and later on, by mid of thirty, she unfortunately lost both eyes vision (No Perception of Light).
The poor visual acuity early live of those patients can be attributed with Macular degeneration. Study done by Azari et al., has shown ten patients with BBS suffered from maculopathy in beginning of their life showing peripheral retinal degeneration and peripheral pigments as well [7].
Postaxial polydactyl, is considered common reported systemic sign in BBS, it was our guide to discover the syndrome. Polydactyly of toes is presented more common than fingers although, our patient has both upper and lower limb polydactyl but she has had the extra fingers excised at childhood [5]. The prevalence gene mutation of BBS is in BBS1 to BBS18 gene accounts for about 70% - 80% world wide [8] While the prevalence of BBS3, BBS9 genes mutations are found in the Saudi Arabia race [9].
Conclusion
Our case has shown advance retinitis pigmentosa (RP) and Maculopathy in middle-aged women resulting blindness of both eyes in a case of Bardet Biedl Syndrome.
In spite, no definite treatment of BBS, the early diagnose, counseling, control of disease manifestation could suppress a variety of organ complication and subsequently reduce morbidity and mortality.
References
- Pearce WG, Gillan JG, Brosseau L. Bardet-Biedl syndrome and retinitis punctata albescens in an isolated northern Canadian community. Canadian journal of ophthalmology. Can J Ophthalmol. 1984 Apr;19(3):115-8. PubMed PMID: 6733577.
- Beales PL, Elcioglu N, Woolf AS, Parker D, Flinter FA. New criteria for improved diagnosis of Bardet-Biedl syndrome: results of a population survey. J Med Genet. 1999 Jun;36(6):437-46. PubMed PMID: 10874630.
- Cherian MP, Sanna'a NA. Clinical spectrum of Bardet–Biedl syndrome among four Saudi Arabian families. Clin Dysmorphol. 2009 Oct;18(4):188- 94. doi: 10.1097/MCD.0b013e32832e4657. PubMed PMID: 19707123.
- Green JS, Parfrey PS, Harnett JD, Farid NR, Cramer BC, Johnson G, et al. The cardinal manifestations of Bardet-Biedl syndrome, a form of Laurence– Moon–Biedl syndrome. N Engl J Med. 1989 Oct 12;321(15):1002-9. PubMed PMID: 2779627.
- Beales PL, Elcioglu N, Woolf AS, Parker D, Flinter FA. New criteria for improved diagnosis of Bardet-Biedl syndrome: results of a population survey. J Med Genet. 1999 Jun;36(6):437-46. PubMed PMID: 10874630.
- Schachat AP, Maumenee IH. Bardet-Biedl syndrome and related disorders. Arch Ophthalmol. 1982 Feb;100(2):285-8. PubMed PMID: 7065946.
- Azari AA, Aleman TS, Cideciyan AV, Schwartz SB, Windsor EA, Sumaroka A, et al. Retinal disease expression in Bardet-Biedl syndrome-1 (BBS1) is a spectrum from maculopathy to retina-wide degeneration. Invest Ophthalmol Vis Sci. 2006 Nov;47(11):5004-10. PubMed PMID: 17065520.
- M'hamdi O, Ouertani I, Chaabouni-Bouhamed H. Update on the genetics of bardet-biedl syndrome. Mol Syndromol. 2014 Feb;5(2):51-6. doi: 10.1159/000357054. Epub 2013 Dec 20. PubMed PMID: 24715851.
- Safieh LA, Aldahmesh MA, Shamseldin H, Hashem M, Shaheen R, et al. Clinical and molecular characterisation of Bardet–Biedl syndrome in consanguineous populations: the power of homozygosity mapping. J Med Genet. 2010 Apr;47(4):236-41. doi: 10.1136/jmg.2009.070755. Epub 2009 Oct 26. PubMed PMID: 19858128.





