Drug Design of Mitochondria-Targeted Antioxidants, Action, Metabolism and Perspectives for Ophthalmic Therapeutics: N-acetylcarnosine Codrug Treatment Platform
Babizhayev MA*
Innovative Vision Products, Inc. Silverside Road, Suite, County of New Castle, Delaware, USA.
*Corresponding Author
Mark A. Babizhayev,
Innovative Vision Products, Inc. , 3511 Silverside Road,
Suite 105, County of New Castle, Delaware USA 19810.
E-mail: markbabizhayev@yahoo.com
Received: March 14, 2017; Accepted: April 17, 2017; Published: April 22, 2017
Citation: Babizhayev MA (2017) Drug Design of Mitochondria-Targeted Antioxidants, Action, Metabolism and Perspectives for Ophthalmic Therapeutics: N-acetylcarnosine Codrug Treatment Platform. Int J Ophthalmol Eye Res. 5(4), 287-307. doi: dx.doi.org/10.19070/2332-290X-1700062
Copyright: Babizhayev MA© 2017. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
Abstract
Maintaining the redox balance within the mitochondria is critical for cellular homeostasis in the eye since the mitochondria host the energy producing systems of the cell and it is widely recognized that damage to the mitochondria plays a key role in sight threatening age-related eye disorders, including retinopathies (maculodystrophy, retinitis pigmentosa , hereditary optic neuropathy), as well as glaucoma, cataract, and autoimmune uveitis.
Reactive oxygen species (ROS) are generated as by-products of cellular metabolism, primarily in the mitochondria. Although ROS are essential participants in cell signaling and regulation, when their cellular production overwhelms the intrinsic antioxidant capacity, damage to cellular macromolecules such as DNA, proteins, and lipids ensues.
Oxidized phospholipids play an important role in execution of the mitochondrial stage of apoptosis and clearance of apoptotic cells. During the lipid peroxidation (LPO) reaction, lipid hydroperoxides are formed as primary products. Several lines of evidence suggest that lipid hydroperoxides can trigger cell death in many cell types, which may be mediated by mitochondria dysfunction pathway. Recently, there was a breakthrough in mitochondrial targeting of antioxidants. Mitochondrial function can be manipulated selectively by targeting bioactive compounds to mitochondria in living cells. Lipophylic antioxidants were covalently coupled to a triphenylphosphonium cation, and these compounds were preferentially taken up by mitochondria.
In this work we proposed the combined use of mitochondria-targeted antioxidant mito Vit E and N-acetylcarnosine, an ophthalmic prodrug of L-carnosine in patented formulation of eye drops including the mucoadhesive compound carboxymethylcellulose to help elucidate the role of mitochondrial oxidative damage in apoptotic cell death. We suggest that mitochondrial oxidative damage plays an important role in ROS-induced apoptosis. Further work using these and other mitochondrially targeted compounds to dissect out the role of mitochondrial oxidative changes in peroxide-induced apoptosis is ongoing.
The findings reported demonstrate that mitochondrially targeted antioxidants such as mito vit E + N-acetylcarnosine in the eye drop formulation with carboxymethylcellulose can be used to investigate the role of mitochondrial oxidative stress in retinal neuronal cells (RGCs) death and may represent a new pharmacologic tool to mitigate complex ocular pathology for the treatment of sight-threatening eye diseases and, especially, neurodegeneration originating from an oxidative injury and glaucomatous neurodegeneration. This strategy also has potential for unraveling the contribution of oxidative stress to other ocular pathologies involving mitochondrial dysfunction.
2.Introduction
3.Oxidation Reactions in Ocular Pathologies
4.Fundamental Role of Mitochondria in Ocular Tissue Metabolism and Ocular Disease Onset (Cataract, Retinal Disease and Glaucoma Onset)
5.The Mitochondria-Targeted Drugs including Mitoquinone (Mito-Q) and Mitovitamin E (Mito-Vit-E) are a New Class of Antioxidants containing the Triphenylphosphonium Cation Moiety that Facilitates Drug Accumulation in Mitochondria
6.The Effects of L-Carnosine on Mitochondria
7.Attractive Targets and Therapeutic Strategies of Drug Delivery to Mitochondria in the Treatment of Oxidative Injury During Glaucomatous Neurodegeneration
8.Acknowledgements
9.Conflict Of Interest
10.References
Keywords
Oxidation-Reduction Mechanisms in the Eye; Ocular Pathologies: Retinopathies (Maculodystrophy, Retinitis Pigmentosa, Hereditary Optic Neuropathy), Glaucoma, Cataract, Autoimmune Uveitis; Mitochondrial Dysfunction and Oxidative Damage; Reactive Oxygen Species Generation; Lipid Peroxidation and Phospholipid Hydroperoxides; Ophthalmic Formulations of N-Acetylcarnosine and Lipophilic Mitochondria-Targeted Antioxidants.
Introduction
Loss of vision is the second greatest, next to death, fear among the elderly. The eye is a unique organ because of its constant exposure to radiation, atmospheric oxygen, environmental chemicals and physical abrasion. The number of people with visual impairment worldwide in 2002 was in excess of 161 million, of whom about 37 million were blind [1]. The burden of visual impairment is not distributed uniformly throughout the world: the least developed regions carry the largest share. Visual impairment is also unequally distributed across age groups, being largely confined to adults 50 years of age and older. A distribution imbalance is also found with regard to gender throughout the world: females have a significantly higher risk of having visual impairment than males. Notwithstanding the progress in surgical intervention that has been made in many countries over the last few decades, cataract remains the leading cause of visual impairment in all regions of the world, except in the most developed countries. Other major causes of visual impairment are, in order of importance, glaucoma, age-related macular degeneration, diabetic retinopathy and trachoma [1].
Oxidation Reactions in Ocular Pathologies
Oxidation-reduction mechanisms have special importance in the eye. Oxidative damage can result in a number of molecular changes that contribute to the development of glaucoma, cataract, and other eye diseases [2-4]. That oxidative stress mechanisms in ocular tissues have been hypothesized to play a role in diseases such as glaucoma, cataract, uveitis, retrolental fibroplasias, age-related macular degeneration and various forms of retinopathy provides an opportunity for new approaches to their prevention and treatment [2]. There are numerous indications of a crucial role of reactive oxygen species (ROS) in the main age-related ocular pathologies (Figure 1), i.e. retinopathies (maculodystrophy [5, 6], retinitis pigmentosa [7, 8], hereditary optic neuropathy [9]), as well as glaucoma [10, 11], cataract [12-15], and autoimmune uveitis [16, 17].
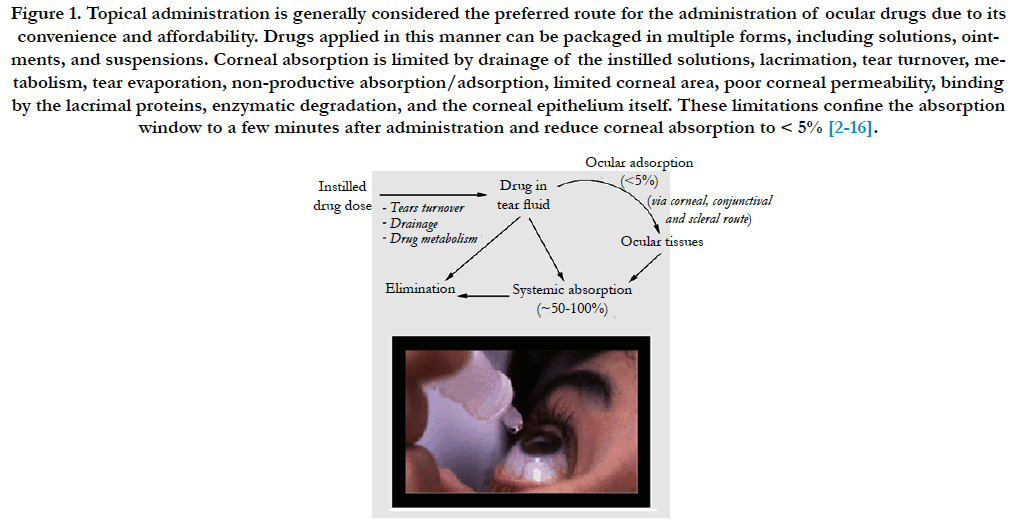
Figure 1. Topical administration is generally considered the preferred route for the administration of ocular drugs due to its convenience and affordability. Drugs applied in this manner can be packaged in multiple forms, including solutions, ointments, and suspensions. Corneal absorption is limited by drainage of the instilled solutions, lacrimation, tear turnover, metabolism, tear evaporation, non-productive absorption/adsorption, limited corneal area, poor corneal permeability, binding by the lacrimal proteins, enzymatic degradation, and the corneal epithelium itself. These limitations confine the absorption window to a few minutes after administration and reduce corneal absorption to < 5% [2-16].
Polyunsaturated fatty acids in mitochondrial cardiolipin are first of all attacked by mitochondria-produced ROS that are quenched by mitochondria-targeted antioxidants [18]. This is why the above-listed pathologies attracted our attention as a possible field of therapeutic application of natural ophthalmic and synthetic mitochondria-targeted antioxidants.
Cataract formation, the opacification of the eye lens, is one of the leading causes of human blindness worldwide, accounting for 47.8% of all causes of blindness [1].
Although great advances have been made in surgical treatment, the incidence of cataracts in developing countries is so high that it overwhelms the capacity of surgical programs.Cataract and age-related macular degeneration (AMD) are the major causes of vision impairment and blindness worldwide. Both conditions are strongly age related with earlier signs (usually asymptomatic) occurring in middle age and becoming severer and more prevalent with increasing age. Primary open-angle glaucoma (POAG), the most common form of glaucoma, is a slowly progressive atrophy of the optic nerve, characterized by loss of peripheral visual function and is usually associated with elevated intraocular pressure. Glaucoma is the second leading cause of vision loss in the world. The number of people with primary glaucoma is estimated at nearly 66.8 million by the year 2000, with 6.7 million of them suffering from bilateral blindness [19]. The molecular basis of POAG is mostly unknown. Several theories of its pathogenesis have been proposed, including mechanic and ischaemic ones (reviewed in ref. [19]). The aetiology of these conditions is thought to fit with the 'free radical theory' of ageing which postulates that ageing and age-related diseases result from the accumulation of cellular damage from ROS. Oxidation–reduction mechanisms have special importance in the eye. Oxidative damage can result in a number of molecular changes that contribute to the development of glaucoma, cataract, and other eye diseases [3, 4]. If the free radical theory of aging is applied to the eye, an altered antioxidant/ oxidant balance should be evident for age-related ocular diseases, such as age-related macular degeneration, cataract, and glaucoma [20].
The studies investigating the relation between POAG, oxidant stress, and antioxidant systems were carried out at the tissue and cellular levels. ROS are generated as by-products of cellular metabolism, primarily in the mitochondria. Although ROS are essential participants in cell signaling and regulation, when their cellular production overwhelms the intrinsic antioxidant capacity, damage to cellular macromolecules such as DNA, proteins, and lipids ensues. Such a state of "oxidative stress" is thought to contribute to the pathogenesis of a number of neurodegenerative diseases. Growing evidence supports the involvement of oxidative stress as a common component of glaucomatous neurodegeneration in different subcellular compartments of retinal ganglion cells (RGCs) (Figure 2). Besides the evidence of direct cytotoxic consequences leading to RGC death, it also seems highly possible that ROS are involved in signaling RGC death by acting as a second messenger and/or modulating protein function by redox modifications of downstream effectors through enzymatic oxidation of specific amino acid residues. Different studies provide cumulating evidence, which supports the association of ROS with different aspects of the neurodegenerative process. Oxidative protein modifications during glaucomatous neurodegeneration increase neuronal susceptibility to damage and also lead to glial dysfunction. Oxidative stress-induced dysfunction of glial cells may contribute to spreading neuronal damage by secondary degeneration. Oxidative stress also promotes the accumulation of advanced glycation end products in glaucomatous tissues. In addition, oxidative stress takes part in the activation of immune response during glaucomatous neurodegeneration, as ROS stimulate the antigen presenting ability of glial cells and also function as co-stimulatory molecules during antigen presentation [11].
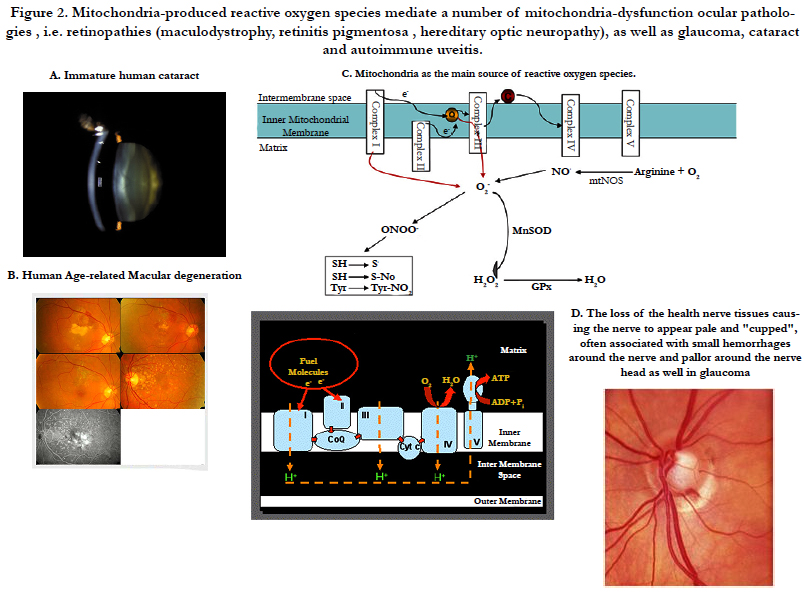
Figure 2. Mitochondria-produced reactive oxygen species mediate a number of mitochondria-dysfunction ocular pathologies , i.e. retinopathies (maculodystrophy, retinitis pigmentosa , hereditary optic neuropathy), as well as glaucoma, cataract and autoimmune uveitis.
Mitochondrial energy production is a major source of endogenous ROS. External sources of ROS include environmental sources especially solar radiation, biomass fuels and tobacco smoking [21-24]. It is estimated that 0.2-2% of oxygen taken up by cells is converted to ROS, through mitochondrial superoxide generation, by the mitochondria. Generation of superoxide at complexes I and III has been shown to occur at both the matrix side of the inner mitochondrial membrane and the cytosolic side of the membrane (Figure 1C). While exogenous sources of ROS such as UV light, visible light, ionizing radiation, chemotherapeutics, and environmental toxins may contribute to the oxidative milieu, mitochondria are perhaps the most significant contribution to ROS production affecting the aging process. In addition to producing ROS, mitochondria are also a target for ROS which in turn reduces mitochondrial efficiency and leads to the generation of more ROS in a vicious self-destructive cycle. Consequently, the mitochondria have evolved a number of antioxidant and key repair systems to limit the damaging potential of free oxygen radicals and to repair damaged proteins [24].
Progressive thinning of the photoreceptor outer nuclear layer resulted from apoptotic cell death. These results support the hypothesis that oxidative damage to the RPE may play a role in some of the key features of AMD [19]. The resulting photooxidative stress can cause acute or chronic retinal damage. The pathogenesis of age-related macular degeneration (AMD), involves oxidative stress and death of the retinal pigment epithelium (RPE) followed by death of the overlying photoreceptors (Figure 3). Evidence suggests that damage due to exposure to light plays a role in AMD and other age-related eye diseases. In the work to King et al., [20], a system for light-induced damage and death of the RPE, based on the human ARPE-19 cell line, was used. Induction of mitochondria-derived reactive oxygen species (ROS) is shown to play a critical role in the death of cells exposed to short-wavelength blue light (425 +/- 20 nm). ROS and cell death are blocked either by inhibiting the mitochondrial electron transport chain or by mitochondria-specific antioxidants. These results show that mitochondria are an important source of toxic oxygen radicals in blue light-exposed RPE cells and may indicate new approaches for treating AMD using mitochondria-targeted antioxidants [20]. In terms of AMD studies, steps in AMD pathogenesis that appear to be good targets for drug development include 1) oxidative damage; 2) lipofuscin accumulation (Figure 4); 3) chronic inflammation; 4) mutations in the complement pathway; and 5) noncomplement mutations that influence chronic inflammation and/or oxidative damage (e.g., mitochondria and extracellular matrix structure). Steps in neovascularization that can be targeted for drug development (Figure 5) and combination therapy include 1) angiogenic factor production; 2) factor release; 3) binding of factors to extracellular receptors (and activation of intracellular signaling after receptor binding); 4) endothelial cell activation (and basement membrane degradation); 5) endothelial cell proliferation; 6) directed endothelial cell migration; 7) extracellular matrix remodeling; 8) tube formation; and 9) vascular stabilization [25].
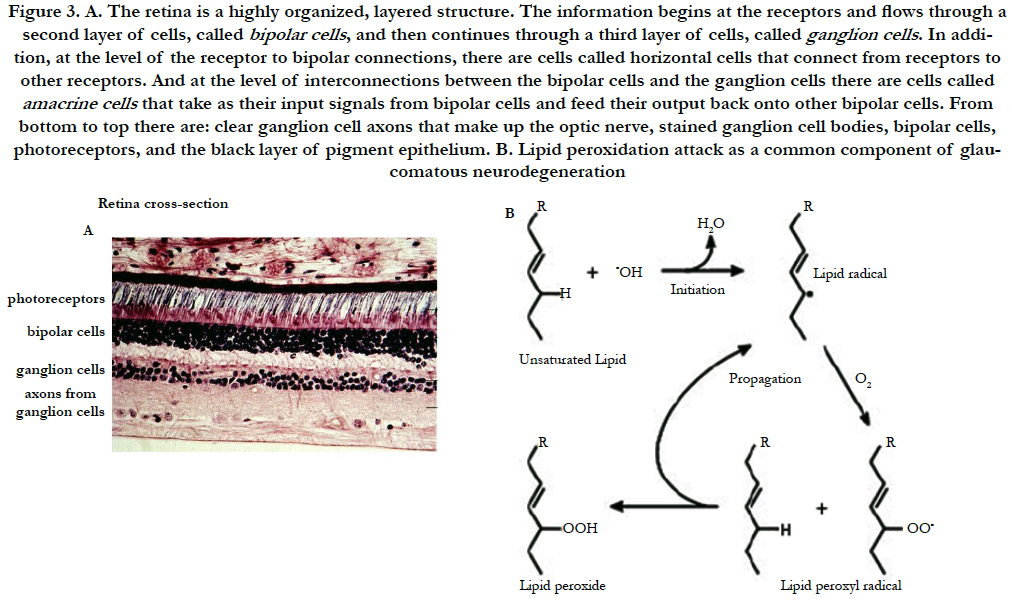
Figure 3. A. The retina is a highly organized, layered structure. The information begins at the receptors and flows through a second layer of cells, called bipolar cells , and then continues through a third layer of cells, called ganglion cells . In addition, at the level of the receptor to bipolar connections, there are cells called horizontal cells that connect from receptors to other receptors. And at the level of interconnections between the bipolar cells and the ganglion cells there are cells called amacrine cells that take as their input signals from bipolar cells and feed their output back onto other bipolar cells. From bottom to top there are: clear ganglion cell axons that make up the optic nerve, stained ganglion cell bodies, bipolar cells, photoreceptors, and the black layer of pigment epithelium. B. Lipid peroxidation attack as a common component of glaucomatous neurodegeneration.
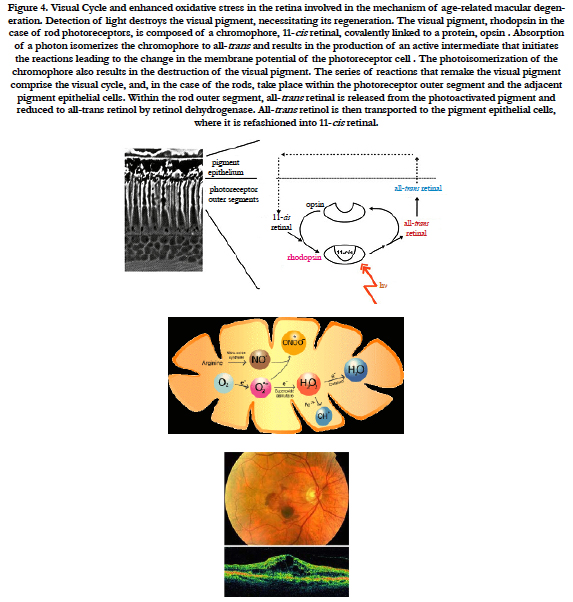
Figure 4. Visual Cycle and enhanced oxidative stress in the retina involved in the mechanism of age-related macular degeneration. Detection of light destroys the visual pigment, necessitating its regeneration. The visual pigment, rhodopsin in the case of rod photoreceptors, is composed of a chromophore, 11-cis retinal, covalently linked to a protein, opsin . Absorption of a photon isomerizes the chromophore to all-trans and results in the production of an active intermediate that initiates the reactions leading to the change in the membrane potential of the photoreceptor cell . The photoisomerization of the chromophore also results in the destruction of the visual pigment. The series of reactions that remake the visual pigment comprise the visual cycle, and, in the case of the rods, take place within the photoreceptor outer segment and the adjacent pigment epithelial cells. Within the rod outer segment, all-trans retinal is released from the photoactivated pigment and reduced to all-trans retinol by retinol dehydrogenase. All-trans retinol is then transported to the pigment epithelial cells, where it is refashioned into 11-cis retinal.
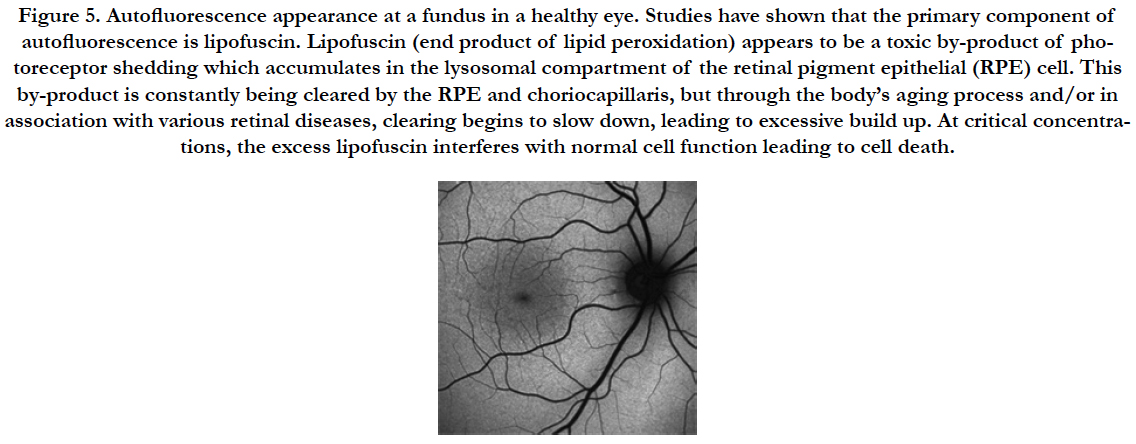
Figure 5. Autofluorescence appearance at a fundus in a healthy eye. Studies have shown that the primary component of autofluorescence is lipofuscin. Lipofuscin (end product of lipid peroxidation) appears to be a toxic by-product of photoreceptor shedding which accumulates in the lysosomal compartment of the retinal pigment epithelial (RPE) cell. This by-product is constantly being cleared by the RPE and choriocapillaris, but through the body’s aging process and/or in association with various retinal diseases, clearing begins to slow down, leading to excessive build up. At critical concentrations, the excess lipofuscin interferes with normal cell function leading to cell death.
Defining the cell biology of choroidal new vessel (CNV) formation and geographic atrophy will lead to identification of different biochemical and cellular pathways that are the target of AMD treatment. Many treatments and treatment combinations are under study for AMD, but all work through a finite number of metabolic pathways. Currently, the most effective proved therapy for AMD-associated CNVs is administered by repeated intravitreal injection of agents that inhibit vascular endothelial growth factor, e.g., ranibizumab. Improved drug delivery will enhance patient satisfaction and possibly will enhance the effectiveness and reduce the risk of current pharmacotherapy for AMD-associated CNVs. Combination therapy (e.g., verteporfin-photodynamic therapy + ranibizumab) appears to reduce the risk and enhance the effectiveness of CNV treatment compared with monotherapy with currently available agents. Improved noninvasive diagnostic imaging may lead to better visual outcomes with existing therapeutic modalities. Improved imaging also may alter favorably the design of future clinical trials for AMD-associated CNVs and thus reduce cost and increase the diversity of sight-saving treatments [26]. Areas of investigation that will advance the field further include combination therapy, improved drug delivery, and improved noninvasive, high-resolution diagnostic imaging. The logistics of future clinical trials will be complicated by the need for an active treatment control group, more stringent definition of successful treatment, and the increased numbers of patients required for combination therapy studies [26].
Importantly, the aging eye appears to be at considerable risk from oxidative stress originated from the potential mitochondrial dysfunction at the certain stages of ophthalmic disease progression and redox balance disturbance in age-related diseases. Mitochondria are critical for ocular function as they represent the major source of a cell's supply of energy and play an important role in cell differentiation and survival.
Mitochondrial dysfunction can occur as a result of inherited mitochondrial mutations (Figure 6) (e.g. Leber's hereditary optic neuropathy and chronic progressive external ophthalmoplegia) or stochastic oxidative damage which leads to cumulative mitochondrial damage and is an important factor in age-related disorders (e.g. age-related macular degeneration, cataract and diabetic retinopathy). Mitochondrial DNA (mtDNA) instability is an important factor in mitochondrial impairment culminating in age-related changes and pathology, and in all regions of the eye mtDNA damage is increased as a consequence of aging and age-related disease. It is now apparent that the mitochondrial genome is a weak link in the defenses of ocular cells since it is susceptible to oxidative damage and it lacks some of the systems that protect the nuclear genome, such as nucleotide excision repair. Accumulation of mitochondrial mutations leads to cellular dysfunction and increased susceptibility to adverse events which contribute to the pathogenesis of numerous sporadic and chronic disorders in the eye [27].
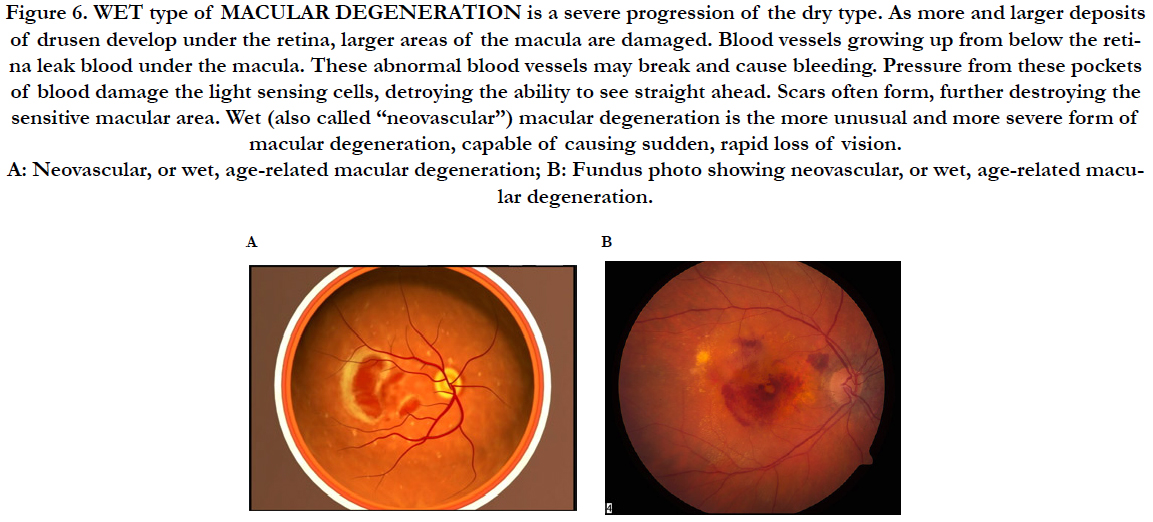
Figure 6. WET type of MACULAR DEGENERATION is a severe progression of the dry type. As more and larger deposits of drusen develop under the retina, larger areas of the macula are damaged. Blood vessels growing up from below the retina leak blood under the macula. These abnormal blood vessels may break and cause bleeding. Pressure from these pockets of blood damage the light sensing cells, detroying the ability to see straight ahead. Scars often form, further destroying the sensitive macular area. Wet (also called “neovascular”) macular degeneration is the more unusual and more severe form of macular degeneration, capable of causing sudden, rapid loss of vision. A: Neovascular, or wet, age-related macular degeneration; B: Fundus photo showing neovascular, or wet, age-related macular degeneration.
Retinitis pigmentosa (RP) is a heterogeneous group of diseases in which one of a wide variety of mutations selectively causes rod photoreceptor cell death (Figure 7). After rods die, cone photoreceptors gradually die resulting in blindness. RP is a label for a group of diseases caused by a large number of mutations that result in rod photoreceptor cell death followed by gradual death of cones [7, 8]. The mechanism of cone cell death is uncertain. Rods are a major source of oxygen utilization in the retina and, after rods die, the level of oxygen in the outer retina is increased. In the study to Komeima et al., [7], the scientists used the rd1 mouse model of RP to test the hypothesis that cones die from oxidative damage. Antioxidants reduce cone cell death in rd1/rd1 mice indicating that cones die from oxidative damage in that model of rapidly progressive RP. A mixture of antioxidants was selected to try to maximize protection against oxidative damage achievable by exogenous supplements; alpha-tocopherol (200 mg/kg), ascorbic acid (250 mg/kg), Mn(III)tetrakis (4-benzoic acid) porphyrin (10 mg/kg), and alpha-lipoic acid (100 mg/kg) [7]. The data support the hypothesis that gradual cone cell death after rod cell death in RP is due to oxidative damage, and that antioxidant therapy may provide benefit [7]. The data suggest that oxidative damage contributes to cone cell death regardless of the disease causing mutation that leads to the demise of rods, and that in more slowly progressive rod degenerations, oxidative damage may also contribute to rod cell death. Protection from oxidative damage may be a broadly applicable treatment strategy in RP [7, 8]. Ghelli et al., [9] have tried to use different paradigms of oxidative and metabolic stress in a cellular model of Leber hereditary optic neuropathy (LHON), with the aim of evaluating the efficacy of potentially therapeutic molecules for the treatment of this disease. Cybrids bearing one of the three most common LHON pathogenic mutations (11778/ND4, 3460/ND1, 14484/ND6) were incubated with two compounds known to induce oxidative injury, tert-butyl hydroperoxide (t-BH) and rotenone. To mimic metabolic stress, cells were incubated in a glucose-free medium containing galactose. Cell viability was determined using the MTT assay. To identify the apoptotic type of cell death, nuclear morphology was examined after cell loading with Hoechst. Cellular glutathione (GSH), and oxidized glutathione (GSSG) levels were measured enzymatically. Incubation with t-BH caused apoptotic cell death of control and LHON cybrids, whereas only LHON cybrids were damaged by rotenone concentrations up to 2.5 muM. Both types of stress caused a marked imbalance in the glutathione levels, but an increase in the GSSG/GSH + GSSG ratio was detected only after rotenone treatment. The efficacy of several antioxidant and antiapoptotic compounds was then assessed in cells exposed to these two oxidative paradigms. Only exogenous GSH remarkably protected the t-BH- and rotenone-treated cybrids from cell death. In contrast, GSH was unable to increase the viability of cybrids exposed to metabolic stress. The results suggest that GSH is an effective antioxidant compound to be tested as a potential treatment for LHON [9]. The era of mitochondria metabolic pathway- based therapy for the early , advanced and late stages of cataract, primary open angle glaucoma (POAG) and AMD has begun [12].
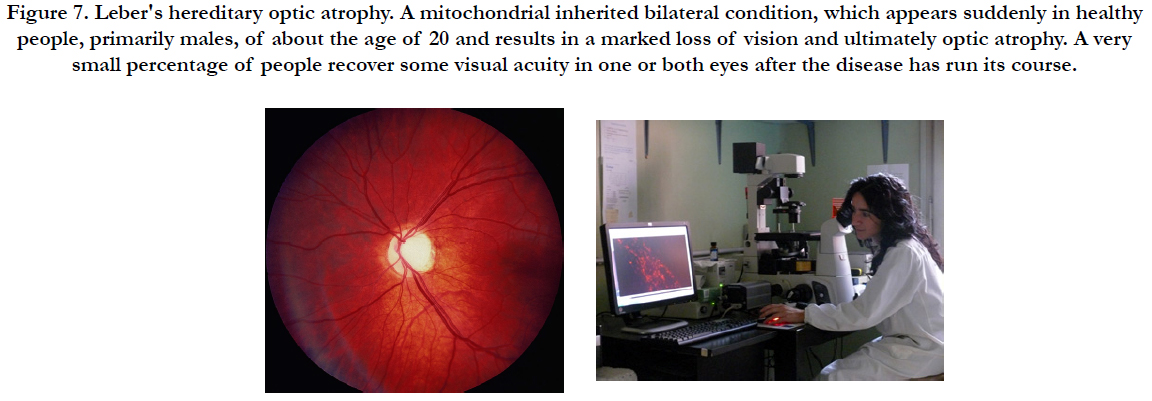

Figure 7. Leber′s hereditary optic atrophy. A mitochondrial inherited bilateral condition, which appears suddenly in healthy people, primarily males, of about the age of 20 and results in a marked loss of vision and ultimately optic atrophy. A very small percentage of people recover some visual acuity in one or both eyes after the disease has run its course.
At each step of the disease development in the free radical oxidation pathway, a new therapeutic treatment could be developed, including topical therapy with patented eye drop formulations but complete inhibition of disease progression will likely require a combination of the various treatments and creation of the specific codrug formulations (Figures 7, 8) [12]. Combination therapy will likely supplant monotherapy as the treatment of choice because the clinical benefits (visual acuity and the specific clinical outcome of treatment) will likely be superior to monotherapy in preventing the early, advanced or even late-stage of cataracts, POAG, AMD and prevention of complications thereof.
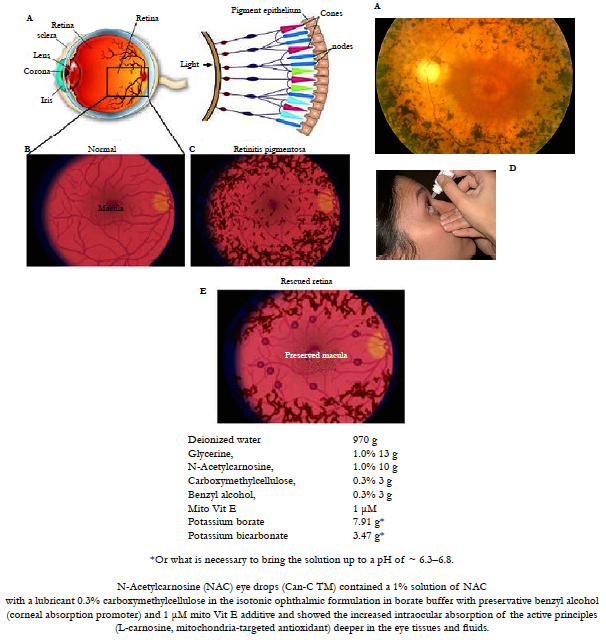

Figure 8. A. Retinitis Pigmentosa (RP) is a group of eye disorders involving progressive degeneration of the retina that leads to severe visual impairment. The age of onset of visuals symptoms is variable from early childhood to adulthood and is usually more severe if the disorder is inherited as an autosomal trait. The disorder usually manifests with decline and loss of night vision during adolescence, followed by loss of peripheral vision in young adulthood, and loss of central vision in later life due to the progressive loss of rod and cone photoreceptors. Common symptoms include night blindness and a decreasing visual field, leading to tunnel vision, legal blindness or, in many cases, complete blindness. Clinical hallmarks are an abnormal fundus with bone-spicule deposits and attenuated retinal vessels. The electroretinogram findings in RP patients show reduced rod and cone response amplitudes. Mutations in the 7 genes analyzed in this panel have been reported in approximately 31%-55% of patients diagnosed with arRP/sporadic RP. Mutations in the CNGA1 gene have been reported in approximately 2%-4% of patients diagnosed with arRP/sporadic RP.
B, C, D, E: Retinitis Pigmentosa is one of the available therapeutic targets for the effective treatment during 4-6 months with the patented combined topical ophthalmic eye drop formulation including mitochondria-targeted antioxidants [27, 104, 105]:
Fundamental Role of Mitochondria in Ocular Tissue Metabolism and Ocular Disease Onset (Cataract, Retinal Disease and Glaucoma Onset)
Mitochondria provide energy generated by oxidative phosphorylation and at the same time play a central role in apoptosis and aging. As a byproduct of respiration, the electron transport chain is known to be the major intracellular site for the generation of ROS [28].
Tissues with high energy demands such as muscles, heart, liver, endocrine glands, brain and retina, have higher numbers of mitochondria per cell. Distribution of mitochondria in the lens is associated with its development. The lens is composed of cells that differentiate from an anterior layer of cuboidal epithelia and migrate posteriorly to form elongated lens fiber cells that make up the lens nucleus [22]. During this process fiber cells synthesize high levels of lens crystallins before losing their nuclei and mitochondria. Thus, the single monolayer of epithelial cells that lines the anterior of the lens, are the only lens cells that carry out aerobic metabolism and contain mitochondria aside from newly differentiated fiber cells. The lens is especially susceptible to damage with aging since lens the cells and their cellular proteins are not turned over or replaced in this encapsulated tissue. The proteins at the center of the eye are some of the oldest in the body and obviously susceptible to age related oxidative damage. Damage to the mitochondria of the epithelial cells may result in ROS production that is thought to affect the proteins of the underlying fiber cells.
Oxidative stress has long been recognized as an important mediator of apoptosis in lens epithelial cells and also plays an important role in the pathogenesis of cataracts [29-33]. The lens exists in an environment that is rich in endogenous sources of ROS, which are produced by the high local oxygen concentration, the chronic exposure to light, and the pathogenic activities of lens epithelial cells [34]. Although multiple physiologic defenses exist to protect the lens from the toxic effects of light and oxidative damage, mounting evidence suggests that chronic exposure to oxidative stress over the long-term may damage the lens and predispose it to cataract development. After exposure to oxidative stress, the redox set point of the single layer of the lens epithelial cells (but not the remainder of the lens) quickly changes, going from a strongly reducing to an oxidizing environment. Almost concurrent with this change is extensive damage to DNA and membrane pump systems, followed by loss of epithelial cell viability and death by necrotic and apoptotic mechanisms. The data suggest that the epithelial cell layer is the initial site of attack by oxidative stress and that involvement of the lens fibers follows, leading to cortical cataract [32]. Lipid compositional changes in lens epithelial cells (HLE B-3) grown in a hyperoxic atmosphere were studied to determine if oxidation could cause changes in the amount and type of phospholipid similar to those found in vivo with age and cataract. The phosphatidylcholines in HLE B-3 cells were 8 times more unsaturated than the sphingomyelins. Cell viability was the same for cells grown for up to 48 h in a normoxic or hyperoxic atmosphere. Lipid peroxidation (LPO) was about three times higher after growth in a hyperoxic atmosphere compared with cells grown in a normoxic atmosphere. The lack of change in the relative amount of sphingomyelin and the decrease in phosphatidylcholine coupled with the increase in lysophosphatidylcholine support the idea that similar mechanisms may be responsible for the lipid compositional changes in both lens epithelial and fiber cells. It is postulated that lipases eliminate oxidized unsaturated glycerolipids, leaving a membrane increasingly composed of more ordered and more saturated sphingolipids. Oxidative stress leads to changes in membrane composition that are consistent with those seen with age in human epithelial cells [34]. Loss of protein sulfhydryl groups, and the oxidation of methionine residues, are progressive and increase as the cataract worsens until > 90% of cysteine and half the methionine residues are oxidised in the most advanced form. By contrast, there may be no significant oxidation of proteins in the centre of the lens with advancing age, even past age 80. The key factor in preventing oxidation seems to be the concentration of nuclear glutathione (GSH) [33].
Apoptosis is a physiologic process of cell death that plays a critical role in a variety of biologic systems, which has been identified as providing an important molecular basis for both the initiation and progression of cataracts [35-37]. There are distinct mechanisms that execute apoptosis according to various different apoptotic stimuli, and these are classified into the mitochondria-dependent pathway (intrinsic pathway) and the death receptor-dependent pathway (extrinsic pathway). Previous studies have demonstrated the capacity of antioxidant protection of the mitochondria-dependent pathway associated with lens opacification in cultured lenses [38-41]. Mitochondrial damage results in the release of cytochrome c from the impaired mitochondria into the cytoplasm, which contributes to programmed cell death [42]. In the rat lenses mitochondrial loss in superficial cortical fibre cells may originate at the sutures and may compensate for loss of optical quality at the sutures [41].
The lens epithelial cells differentiate into lens fiber cells through a process, which utilizes the same regulators as those in apoptosis at multiple signaling steps. Both in vitro and in vivo studies have shown that treatment of adult lens with stress factors induces apoptosis of lens epithelial cells, which is followed by cataractogenesis [35]. It is well established that various factors such as oxidative stress, UV, and other toxic agents can induce both in vivo and in vitro cataract formation. However, a common cellular basis for this induction has not been previously recognized. The published study of lens epithelial cell viability suggests such a general mechanism [37]. When lens epithelial cells from a group of 20 cataract patients 12 to 94 years old were analyzed by terminal deoxynucleotidyl transferase (TdT) labeling and DNA fragmentation assays, it was found that all of these patients had apoptotic epithelial cells ranging from 4.4 to 41.8%. By contrast, in eight normal human lenses of comparable age, very few apoptotic epithelial cells were observed. Li et al., [37] suggest that cataract patients may have deficient defense systems against factors such as oxidative stress and UV at the onset of the disease. Such stress can trigger lens epithelial cell apoptosis that then may initiate cataract development. To test this hypothesis, it has been also demonstrated that hydrogen peroxide at concentrations previously found in some cataract patients induces both lens epithelial cell apoptosis and cortical opacity [37]. Moreover, the temporal and spatial distribution of induced apoptotic lens epithelial cells precedes development of lens opacification. These results suggest that lens epithelial cell apoptosis may be a common cellular basis for initiation of noncongenital cataract formation [37]. Transforming growth factor beta(2) (TGF-beta(2)), a growth regulator of human lens epithelial cells (HLECs), also regulates the death of lens epithelial cells. Dose-response analysis showed that the TGF-beta(2) concentration needed to induce HLECs death (100 pg/ml) was 10 times that needed to inhibit growth in these cells (10 pg/ml) [36]. TGF-beta(2)-induced apoptosis in HLECs was preceded by an induction of ROS and a decrease in glutathione in the intracellular content, indicating that this factor induces oxidative stress in HLECs. Studies performed to analyze the levels of c-fos mRNA, a gene whose expression is modulated by the redox state, demonstrated that only high, apoptotic concentrations of TGF-beta(2) (100 pg/ml) produced an increase in the mRNA levels of this gene, the level of induction being similar to that found when cells were incubated in the presence of hydrogen peroxide [36]. Finally, the cell death induced by TGF-beta(2) in HLECs was partially blocked by radical scavengers, which decreased the percentage of apoptotic cells, whereas these agents did not modify the growth-inhibitory effect elicited by TGF-beta(2) in these cells. The results presented in this published study provide evidence for the involvement of an oxidative process in the apoptosis elicited by TGF-beta(2) in HLECs [36]. The effect of R, S, and racemic forms of α-lipoic acid was tested on the formation of opacity in normal rat lenses incubated with 55.6 mM glucose, as a model for in vivo diabetic cataractogenesis [40]. Control lenses, incubated 8 days with 5.56 mM glucose, did not develop opacities. Formation of lens opacities in vitro was correlated with lactate dehydrogenase (LDH) leakage into the incubation medium. Opacity formation and LDH leakage, resulting from incubation in medium containing 55.6 mM glucose to model diabetes, were both suppressed by the addition of 1 mM R-lipoic acid. Addition of 1 mM racemic lipoic acid reduces these damaging effects to the lens by one-half, while S-lipoic acid potentiated LDH leakage, consistent with the hypothesis that R-lipoic acid is the active form. Although HPLC analysis demonstrated that both stereoisomers of lipoic acid were reduced to dihydrolipoate at comparable rates by the intact lens, the mitochondrial lipoamide dehydrogenase system is highly specific for reduction of exogenous R-lipoic to dihydrolipoic acid. Stereospecific protection against this opacity is consistet with specific reduction of R-lipoic acid in mitochondria of the vulnerable cells at the lens equator where the first globular degeneration is seen in glucose cataract [40].
Exposure to solar and occupational ultraviolet (UV) radiation, and thus production of ROS and subsequent cell death, has been implicated in a large spectrum of skin and ocular pathologies, including cataract. Retinal pigment epithelial cell apoptosis generates photoreceptor dysfunction and ultimately visual impairment. Normal morphological change is presumably allowing energy transmission across the cell from regions of low to regions of high ATP demand. Lack of mitochondrial movement, fragmentation, and swelling of mitochondria may represent early morphological changes following oxidative stress that may lead to activation of caspase-mediated apoptotic pathways [28].
The retina is the most oxygen consuming tissue in the body with consumption level around 50% higher than the brain or kidneys [43]. The central retina mediates high acuity vision, and its progressive dysfunction due to macular degeneration is the leading cause of visual disability among adults in industrialized societies [43]. The macula is a source of high metabolic activity, and is therefore exposed to correspondingly high levels of ROS. With age, the balance between production of ROS and local antioxidant levels is shifted, and damage ensues. Systemic ROS and antioxidant levels in AMD reflect these local processes [44]. In the retina, mitochondria are found throughout but the highest number of mitochondria per cell is found in the photoreceptors. The retina captures and converts light between 400-760 nm into electrical signals that are sent to the brain by way of the optic nerve and in the process helps to translate these electrical signals into what is known as vision. The same light that allows vision to occur is nevertheless also potentially toxic to retinal cells in certain situations. The shorter wavelengths of light are known to interact with chromophores in photoreceptors and pigment epithelial cells to cause oxidative stress and severe damage. Indeed it is generally accepted that short wavelength light effects is one cause for loss of photoreceptor function in age-related macular degeneration. Recent studies have demonstrated that light may be a contributing factor for the death of retinal ganglion cells in certain situations. Light as impinging on the retina, especially the short wavelength form, affect mitochondrial chromophores and can result in neurone death. Importantly ganglion cell axons within the eye are laden with mitochondria and unlike the outer retina are not protected from short wavelength light by macular pigments. It has therefore been proposed that when ganglion cell function is already compromised, as in glaucoma, then light impinging on their mitochondria might be a contributor to their eventual demise [45].
Mitochondria are central to retinal cell function and survival. There is increasing evidence to support an association between mitochondrial dysfunction and a number of retinal pathologies including age-related macular degeneration (AMD), diabetic retinopathy and glaucoma. The past decade has highlighted mitochondrial genomic instability as an important factor in mitochondrial impairment culminating in age-related changes and age-related pathology. This represents a combination of the susceptibility of mitochondrial DNA (mtDNA) to oxidative damage and a limited base excision repair pathway. This random cumulative mtDNA damage leads to cellular heteroplasmy and, if the damage affects a sufficient proportion of mitochondria within a given cell, results in loss of cell function and greater susceptibility to stress.
mtDNA damage is increased in the neural retina and RPE with ageing and appears to be greatest in AMD. It thus appears that the mitochondrial genome is a weak link in the antioxidant defenses of retinal cells and that deficits in mitochondrial DNA (mtDNA) repair pathways are important contributors to the pathogenesis of retinal degeneration [46].
Specifically targeting mitochondria with pharmacological agents able to protect against oxidative stress or promote repair of mtDNA damage may offer potential alternatives for the treatment of retinal degenerations such as AMD [46].
ROS Production in Mitochondria
Mitochondria are sometimes referred to as the powerhouses of the cell since they generate most of the cells chemical energy requirement in the form of adenosine triphosphate (ATP). They also have a significant role in regulating apoptosis and necrosis, ROS levels, cellular signaling, control of the cell cycle, and growth and differentiation [47]. The new awareness has sparked a new and growing area of mitochondrial research, that has become of great interest to a wide variety of scientists ranging from those involved in elucidating the role of mitochondria in cell death and aging to those interested in either suppressing or facilitating these processes as it relates to identifying new therapies or drugs for human disease [47]. Mitochondria are also involved in calcium uptake and release, production of NADH, synthesis of DNA, RNA and proteins, DNA repair and metabolic pathways.
Mitochondria are a double membrane organelle with four distinct compartments, the outer membrane, inner membrane, intermembrane space and the matrix. It is the only organelle apart from the nucleus to contain its own DNA, mtDNA is a circular molecule of just over 16000 base pairs making up 37 genes that encode 13 components of the electron transport chain, and transcription and translational machinery [22]. Oxidation of fuels, such as glucose, generates reducing equivalents that feed into the electron transport chain of the mitochondria [48]. The electron transport chain generates ATP by oxidative phosphorylation, creating a proton gradient through sequential transfer of electrons donated by reducing equivalents. This complex system is made up of five multi enzyme subunits, NADH dehydrogenase comprises complex I (46 subunits), succinate dehydrogenase is complex II (4 subunits), cytochrome C reductase and cytochrome C oxidase make up complexes III and IV respectively (11 and 13 subunits). Complex V is the ATP synthase (16 subunits) that uses the proton gradient created by the first four complexes to drive phosphorylation of ADP to form the energy rich ATP. A great deal of research indicates that dysfunctional mitochondria are the primary site of ROS [49]. In addition to the well-established role of the mitochondria in energy metabolism, regulation of cell death has recently emerged as a second major function of these organelles. This, in turn, seems to be intimately linked to their role as the major intracellular source of ROS which are mainly, generated at Complex I and III of the respiratory chain. Excessive ROS production can lead to oxidation of macromolecules and has been implicated in mtDNA mutations, ageing, and cell death. Although mitochondrial dysfunction can cause ATP depletion and necrosis, these organelles are also involved in the regulation of apoptotic cell death by mechanisms, which have been conserved through evolution. The essential role of mitochondrial oxidative phosphorylation in cellular energy production, the generation of reactive oxygen species, and the initiation of apoptosis has suggested a number of novel mechanisms for mitochondrial pathology [50]. Increasingly, mitochondria are thought to play a regulatory role in cell death partly due to its role as a source of ROS and due to the release of cytochrome C and other pro-apoptotic factors that activate caspases and trigger apoptosis [22]. Cytochrome c release occurs by a two-step process that is initiated by the dissociation of the hemoprotein from its binding to cardiolipin, which anchors it to the inner mitochondrial membrane. Oxidation of cardiolipin reduces cytochrome c binding and results in an increased level of "free" cytochrome c in the intermembrane space. Conversely, mitochondrial antioxidant enzymes protect from apoptosis [51].
Cytochrome C is a small globular heme containing electron carrier in the electron transport chain of the mitochondria. Its primary role in the electron transport chain is a crucial one, shuttling electrons from Complex III (ubiquinol: cytochrome c reductase) to complex IV (cytochrome oxidase), however, its release from the mitochondria to the cytosol is the initiating factor for the internal apoptotic pathway. The release of cytochrome C is a two step process, initiated by release of the hemoprotein from its binding to cardiolipin at the inner mitochondrial membrane [51]; this results in a pool of free cytochrome C in the intermembrane space. Subsequent permeabilization of the outer mitochondrial membrane releases cytochrome C into the cytosol where it binds apoptotic peptidase activating factor 1 (APAF1) [22]. Many lethal agents target the mitochondria and cause release of cytochrome c and other pro-apoptotic proteins, which can trigger caspase activation and apoptosis [49]. Conversely, mitochondrial antioxidant enzymes protect from apoptosis [51].
Mitochondria are a cell's single greatest source of ROS. ROS are important for many life sustaining processes of cells and tissues, but they can also induce cell damage and death. If their production and levels within cells is not effectively controlled, then the detrimental effects of oxidative stress can accumulate. Oxidative stress is widely thought to underpin many ageing processes, and the oxidative stress theory of ageing is one of the most widely acknowledged theories of ageing. As well as being the major source of ROS, mitochondria are also a major site of oxidative damage [48]. More than 95% of O2ˉ•, produced during normal metabolism is generated by the electron transport chain in the inner mitochondrial membrane. Correspondingly, mitochondria are also the major target of ROS [49]. Such damage to the mitochondrial DNA may contribute to the age-associated decline in mitochondrial function. Mitochondrial DNA may be more susceptible to oxidative damage than to nuclear DNA [52] because of its proximity to electron transport chains, lack of protective histones, and insufficient repair mechanisms [53]. Furthermore, mitochondrial DNA encodes several subunits of different respiratory complexes [50]. ROS-induced DNA damage may lead to the production of more ROS, thus perpetuating a vicious cycle.
Hence, there is accumulating evidence supporting a direct link between mitochondria, oxidative stress and cell death.
The Mitochondria-Targeted Drugs including Mitoquinone (Mito-Q) and Mitovitamin E (Mito- Vit-E) are a New Class of Antioxidants containing the Triphenylphosphonium Cation Moiety that Facilitates Drug Accumulation in Mitochondria
Enhanced mitochondrial oxidative damage is a prominent feature of most age-related human diseases including human cataract and neurodegenerative eye disorders, such as age-related macular degeneration and glaucomatous neuropathy. Aberrant electron leakage from mitochondria in the respiratory chain in oxidant-stressed cells triggers the formation of ROS [54], leading to enhanced oxidative damage in mitochondria [54]. The increased oxidative damage in mitochondria induces the mitochondrial permeability transition resulting in apoptotic or necrotic cell death [55]. Oxidants generated by mitochondria appear to be the major source of the oxidative lesions that accumulate with age. Several mitochondrial functions decline with age. The contributing factors include the intrinsic rate of proton leakage across the inner mitochondrial membrane (a correlate of oxidant formation), decreased membrane fluidity, and decreased levels and function of cardiolipin, which supports the function of many of the proteins of the inner mitochondrial membrane. Age-associated accumulation of mitochondrial deficits due to oxidative damage is likely to be a major contributor to cellular, tissue, and organismal aging [55]. Mitochondria play a major role in regulating apoptosis through enhanced release of cytochrome c that results in the activation of caspases and subsequent cell death [56, 57]. Alterations in mitochondrial membrane structure and function can occur in a caspase-independent fashion and have a higher predictive value for cell death than caspase activation. Douglas Green and Guido Kroemer argue that caspases might have a dual function in the apoptotic process: first, as signal-transduction molecules that act as facultative inducers of mitochondrial membrane changes, and, second, as processing enzymes that orchestrate the apoptotic phenotype. They propose a model for initiation of apoptosis in which mitochondria and caspases engage in a self-amplifying pathway of mutual activation [56].
The essential role of mitochondrial oxidative phosphorylation in cellular energy production, the generation of ROS, and the initiation of apoptosis has suggested a number of novel mechanisms for mitochondrial pathology in ophthalmic diseases [12, 22]. Glaucoma is increasingly recognized as a neurodegenerative disorder, characterized by the accelerated loss of retinal ganglion cells (RGCs) and their axons. Open angle glaucoma prevalence and incidence increase exponentially with increasing age, yet the pathophysiology underlying increasing age as a risk factor for glaucoma is not well understood. Accumulating evidence points to age-related mitochondrial dysfunction playing a key role in the etiology of other neurodegenerative disorders including amyotrophic lateral sclerosis, Alzheimer and Parkinson disease. The 2 major functions of mitochondria are the generation of ATP through oxidative phosphorylation and the regulation of cell death by apoptosis. Kong et al., [58] have summarized the evidence to support the hypothesis that age-associated mitochondrial dysfunction renders RGCs susceptible to glaucomatous injury by reducing the energy available for repair processes and predisposing RGCs to apoptosis. Eliciting the role of mitochondria in glaucoma pathogenesis may uncover novel therapeutic targets for protecting the optic nerve and preventing vision loss in glaucoma [58]. Aging is the greatest risk factor for glaucoma, implying that intrinsic age-related changes to retinal ganglion cells, their supporting tissue or both make retinal ganglion cells susceptible to injury. Changes to the ocular vasculature, connective tissue of the optic nerve head and mitochondria, which have been documented with advancing age and shown to be exacerbated in glaucoma, may predispose to glaucomatous injury. When considering such age-related changes, it is difficult to separate pathological change from physiological change, and cause from consequence. The insults that predispose aged retinal ganglion cells to injury are likely to be varied and multiple; therefore, it may be more relevant to identify and treat common mechanisms that predispose to retinal ganglion cell failure and/or death. Chrysostomou et al., [59] have suggested that mitochondrial dysfunction, as either a cause or consequence of injury, renders retinal ganglion cells sensitive to degeneration. Therapeutic approaches that target mitochondria and promote energy production may provide a general means of protecting aged retinal ganglion cells from degeneration, regardless of the etiology [59].
Mitochondrial abnormality has been implicated in various models of RGC degeneration. Munemasa et al., [60] investigated modulation of mitochondrial membrane permeability and apoptosis-inducing factor (AIF) translocation in a rat experimental glaucoma model. A decrease in MitoTracker-labeled mitochondria around the lamina area of the optic nerve was observed in the glaucomatous eye. Immunoblot analysis for axonal motor proteins showed that a significant decrease in kinesin 1 and myosin Va levels in the glaucomatous optic nerve. A significant decrease in mitochondrial thioredoxin 2 (Trx2) level was observed in the optic nerve after intraocular pressure (IOP) elevation. Translocation of AIF from the mitochondria to the axoplasm and nucleus was observed in the axon and cell body, respectively. Trx2 over-expression in the mitochondrial membrane of RGC-5 cells inhibited AIF translocation, resulting in cytoprotective effect against neurotoxicity induced by TNF-α/buthionine sulfoximine treatment. in vivo transfection was performed with EGFP-Trx2 plasmid and electroporation. Over-expression of Trx2 in the retina and optic nerve indicated the protective effect against high IOP induced axonal degeneration. Thus, the decreased mitochondrial membrane potential and subsequent AIF translocation were involved in the glaucomatous neurodegeneration. Furthermore, modulation of mitochondria through the inhibition of AIF translocation may become a new treatment strategy for neurodegenerative disease, such as glaucoma [60]. Mitochondria are critical for ocular function as they represent the major source of a cell's supply of energy and play an important role in cell differentiation and survival [60].
Several systemic or neurological mitochondrial diseases, including amyotrophic lateral sclerosis, Parkinson’s, Alzheimer’s, and Huntington’s diseases, are characterized by dysfunctional or defective respiratory chain components (e.g. complexes I and III) that exacerbate ROS formation [50, 55, 58, 61-68]. In addition, levels of mitochondrial iron increased under these pathological conditions [63-67] . Glutathione (GSH) exhibits several functions in the brain chiefly acting as an antioxidant and a redox regulator. GSH depletion has been shown to affect mitochondrial function probably via selective inhibition of mitochondrial complex I activity [63]. Replenishment of normal glutathione levels within the brain may hold an important key to therapeutics [63, 64].
The selective mitigation of oxidative damage in mitochondria is therefore an effective therapeutic strategy in such age-related human disorders. However, a major limitation of antioxidant therapy in the treatment of mitochondrial diseases has been the inability to enhance antioxidant levels in mitochondria [68]. Strategies to prevent mitochondrial damage or to manipulate mitochondrial function in clinically useful ways may provide new therapies for a range of human ocular disorders. We have outlined why mitochondria are a potentially important target for drug delivery and herewith we discuss how to deliver bioactive molecules selectively to mitochondria within cells during the treatment of ophthalmic disorders [12]. Recently, there was a breakthrough in mitochondrial targeting of antioxidants [12, 69, 70]. Mitochondrial function can be manipulated selectively by targeting bioactive compounds to mitochondria in living cells [69, 70]. Lipophylic antioxidants were covalently coupled to a triphenylphosphonium cation, and these compounds were preferentially taken up by mitochondria (Figure 8) [12, 69, 70]. These agents initially accumulated in the cytoplasmic region of cells because of the negative plasma membrane potential (30–60 mV) [12, 69-71]. The lipophilic cations easily permeate through the lipid bilayers and subsequently accumulate several hundred-fold within mitochondria because of a large mitochondrial membrane potential (150–170 mV; negative inside). These compounds pass easily through all biologic membranes, including the ocular–blood barrier, and thus reach those ocular tissues most affected by mitochondrial oxidative damage. Murphy and coworkers initiated the practical realization of the theoretical concept of mitochondria-targeted antioxidants [68, 69, 72, 73]. They synthesized and tested just a few of mitochondria- targeted antioxidants conjugated to the lipophilic alkyltriphenylphosphonium cations. Mito-Q, a derivative of ubiquinone, and MitoVit-E, a derivative of Vit-E, are two promising antioxidants (Figure 8) that are specifically targeted to mitochondria [12, 69-75]. Mitochondrial ubiquinone is a respiratory chain component buried within the lipid core of the inner membrane where it accepts 2 electrons from complexes I or II forming the corresponding reduction product i.e. ubiquinol) which then donates electrons to complex III [76]. The ubiquinone pool in vivo exists largely in the reduced ubiquinol form acting as an antioxidant and a mobile electron carrier. alpha-Tocopherol (vitamin E) is a lipophilic chain-breaking antioxidant which inhibits lipid peroxidation in isolated mitochondrial membranes and protects membranes from oxidative damage. The primary oxidation product of vitamin E is the tocopheroxyl radical. By using submitochondrial particle membranes, it was shown that NADH, succinate, and reduced cytochrome c-linked oxidation reduce the tocopheroxyl radical, preventing both accumulation of the radical and vitamin E consumption. As the electron transport chain can reduce tocopheroxyl radical it may have an important physiological role in recycling vitamin E [78].
Ubiquinol has been reported to function as an antioxidant by donating a hydrogen atom from one of its hydroxyl groups to a lipid peroxyl radical, thereby decreasing lipid peroxidation (LPO) within the mitochondrial inner membrane [77-79]. The ubisemiquinone radical formed during this process disproportionates into ubiquinone and ubiquinol [80, 81]. The respiratory chain subsequently recycles ubiquinone back to ubiquinol, restoring its antioxidant function. Vitamin E (Vit-E) (or α-tocopherol) is another antioxidant within the mitochondrial inner membrane, and the tocopheroxyl radical formed from one electron oxidation of Vit-E regenerates Vit-E by reacting with ubiquinol [75, 77, 80, 82]. The objective of the published study to Lass and Sohal [75] was to elucidate the anti-oxidative roles of coenzyme Q (CoQ) and alpha-tocopherol in mitochondrial membranes by determining whether CoQ directly scavenges peroxyl- and alkoxyl-radicals or indirectly regenerates alpha-tocopherol during autooxidation of mitochondrial membranes. A comparison of the interaction between alpha-tocopherol and CoQ during autooxidation was made between bovine and rat heart mitochondria, which differ approximately 15-fold in their alpha-tocopherol content. Results of this study indicate that during autooxidation of mitochondria, alpha-tocopherol acts as the direct radical scavenger, whereas ubiquinol regenerates alpha-tocopherol [75].
Results suggest that mitochondria-targeted antioxidants are more effective inhibitors of mitochondrial damage and apoptosis than the corresponding “untargeted” counterparts (i.e. Vit-E) (Figures 8,9 ) [61].
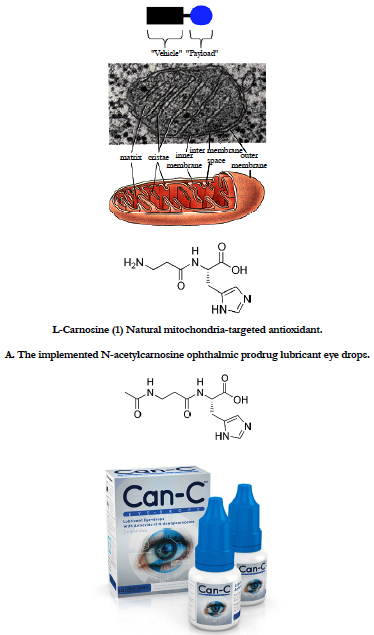
Figure 9. Concept of targeting mitochondria with functional agents [1-9] that use a vehicle to deliver an ROS scavenging payload into mitochondria. With the recognition of the central role of mitochondria in apoptosis, there is a need to develop specific tools to manipulate mitochondrial function within cells. Here we report on the development of a novel antioxidant that selectively blocks mitochondrial oxidative damage, enabling the roles of mitochondrial oxidative stress in different types of cell death to be inferred. For details, see section 5 in the text.
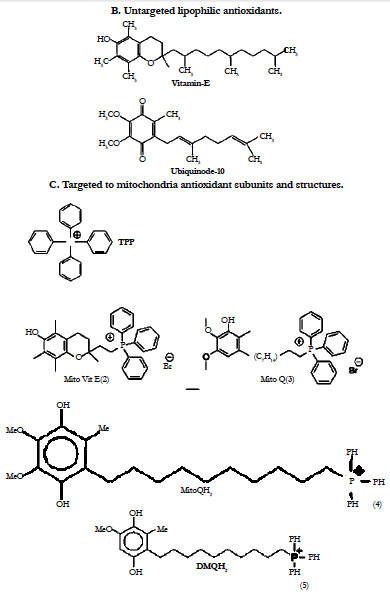
Figure 9. Concept of targeting mitochondria with functional agents [1-9] that use a vehicle to deliver an ROS scavenging payload into mitochondria. With the recognition of the central role of mitochondria in apoptosis, there is a need to develop specific tools to manipulate mitochondrial function within cells. Here we report on the development of a novel antioxidant that selectively blocks mitochondrial oxidative damage, enabling the roles of mitochondrial oxidative stress in different types of cell death to be inferred. For details, see section 5 in the text.
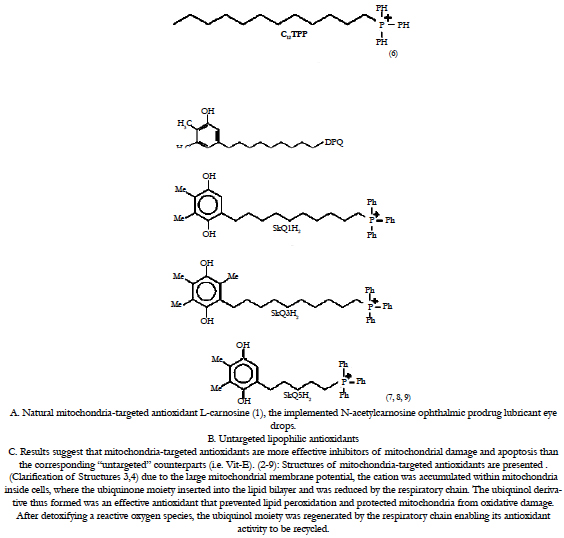
Figure 9. Concept of targeting mitochondria with functional agents [1-9] that use a vehicle to deliver an ROS scavenging payload into mitochondria. With the recognition of the central role of mitochondria in apoptosis, there is a need to develop specific tools to manipulate mitochondrial function within cells. Here we report on the development of a novel antioxidant that selectively blocks mitochondrial oxidative damage, enabling the roles of mitochondrial oxidative stress in different types of cell death to be inferred. For details, see section 5 in the text.
Figure 10. N-acetylcarnosine as the ophthalmic prodrug of the natural mitochondria-targeted antioxidant L-carnosine in the topical ophthalmic application.
In 2006, an attempt was undertaken in Skulachev’s group to replace the ubiquinone moiety in MitoQ by plastoquinone. As a result, a series of mitochondria-targeted antioxidants named SkQ has been synthesized (Figure 8) [83, 84]. It was reported [85-87] that the reactivity of the ‘‘tailless’’ plastoquinol analogs to the peroxyl radicals was higher than that of natural ubiquinols.
Oxidized phospholipids play an important role in execution of the mitochondrial stage of apoptosis and clearance of apoptotic cells. During the LPO reaction, lipid hydroperoxides are formed as primary products. Several lines of evidence suggest that lipid hydroperoxides can trigger cell death in many cell types, which may be mediated by mitochondria dysfunction pathway. With extended hyperoxic insult, the oxidants overwhelm the antioxidant defense system and eventually cell death ensues [88]. ROS generation correlated inversely with mitochondrial membrane potential and the amount of cardiolipin, factors likely to contribute to loss of cell viability [88]. The critical role of mitochondria in programmed cell death leads to the design of mitochondriotropic agents as a strategy in regulating apoptosis. According to the new IVP mitochondrion concept, mitochondria do not exist stably as distinct, individual, autonomous organelles. Rather, mitochondria form a network within cells; their continous fusion and fission is a highly dynamic process, adapting to the role the mitochondrion actually has in the cell. Increasing results confirm the role of mitochondrial fission and fragmentation in most forms of apoptosis, even as a cause. This suggests that fragmented mitochondria are in a „bad“ condition, under oxidative stress. To overcome this problem, we developed natural forms of mitochondria-targeted antioxidants (Figure 8, Structure (1)), non-hydrolized carnosine associated with the ophthalmic carrier typified for topical use (N-acetylcarnosine lubricant eye drops) (major proposed administration way), ocular injection (intra-vitreal, subconjunctival, parabulbar) or even oral administration (synergistic administration with chaperones and reduced glutathione synthesis booster) [89-92].
The Effects of L-Carnosine on Mitochondria
The effect of carnosine on self-organization of mitochondrial assemblies was studied in rat liver homogenate of quiescent and excited animals. It was shown in separate electron microscopy experiments with serial slices that under our conditions of preparation of homogenate, blocks of native mitochondrial-reticular net work in the cell, assemblies of mitochondria, are kept. Carnosine was shown to prevent dissociation of assemblies during storage. Its effect is maximal for more dissociated assemblies from excited animals with decreased ability for self-organization. Prevention of disassembly of organelles by carnosine can serve as one of the mechanisms of carnosine-induced diminishing of muscle fatigue under prolonged work [93]. L-Carnosine prevented both 12-O-tetradecanoylphorbol-13-acetate (TPA)-and H2O2-induced DNA fragmentation, the loss of mitochondrial membrane potentials and blocked the release of cytochrome c into cytosol. Subsequently, the cleavages of poly (ADP-ribose) polymerase were significantly reduced in L-carnosine-treated cells. However, western blotting analysis revealed that p53 protein level did not change for 12h after TPA- and H2O2-treatment. Therefore, these results suggested that L-carnosine, an antioxidant, protected both H2O2- and TPA-induced apoptosis through mitochondrial pathways [94]. Carnosine addition to mitochondria or its accumulation in mitochondria under hypoxia is associated with activation of alpha-ketoglutarate oxidation and its formation through transamination [95].
Recently, phospholipid peroxidation products gained a reputation as key regulatory molecules and participants in oxidative signaling pathways. Oxidation of two anionic phospholipids--cardiolipin (CL) in mitochondria and phosphatidylserine (PS) in extramitochondrial compartments--is important signaling event, particularly during the execution of programmed cell death and clearance of apoptotic cells. Quantitative analysis of CL and PS oxidation products is central to understanding their molecular mechanisms of action [96, 97]. Furthermore, specific characteristics of CL in mitochondria--its asymmetric transmembrane distribution and mechanisms of collapse, the regulation of its synthesis, remodeling, and fatty acid composition--are given significant consideration [97, 98]. Cytochrome c (cyt c) acts as a CL-specific peroxidase very early in apoptosis. At this stage, the hostile events are still secluded within the mitochondria and do not reach the cytosolic targets. CL oxidation process is required for the release of pro-apoptotic factors into the cytosol. Manipulation of cyt c interactions with CL, inhibition of peroxidase activity, and prevention of CL peroxidation are prime targets for the discovery of anti-apoptotic drugs acting before the "point-of-no-return" in the fulfillment of the cell death program. During apoptosis, a mitochondria- specific phospholipid, cardiolipin (CL), interacts with cytochrome c (cyt c) to form a peroxidase complex that catalyzes CL oxidation; this process plays a pivotal role in the mitochondrial stage of the execution of the cell death program. Several works were focused on redox mechanisms and essential structural features of cyt c's conversion into a CL-specific peroxidase that represent an interesting and may be still unique example of a functionally significant ligand change in hemoproteins [96, 97]. Recently, it was demonstrated that peroxidase cyt c/CL complexes can utilize free fatty acid hydroperoxides (FFA-OOH) at exceptionally high rates that are approximately 3 orders of magnitude higher than for H2O2 [96]. Interaction of cyt c with negatively charged lipid membranes induces considerable disruption of the native compact structure of the protein and induces intermediate conformations between the native and the fully unfolded states, called a “molten globule.” This state, an “alternative folding,” is defined as a compact conformation with a secondary structure comparable to that of the native state and fluctuating tertiary conformation due to a high enhancement of intramolecular motion [99-102]. Phosphorus-31 NMR has been used to investigate the interaction of cytochrome c with bilayers of the anionic lipids dioleoyl phosphatidylglycerol (DOPG), dioleoyl phosphatidylserine (DOPS), and diacyl phosphatidylinositol (diacylPI). All 31P NMR spectra revealed the typical line shapes characteristics of phospholipids in liquid-crystalline bilayers. The effects on the 31P chemical shift anisotropy (CSA) for each system reflect particular modes of phospholipid headgroup interaction with cytochrome c [99]. The observed destabilization of protein structure mediated by acidic phospholipids (and possibly formation of folding intermediates at the membrane surface) may represent a general property of a larger class of water-soluble proteins for which membrane binding is governed by electrostatic forces [101]. In solution, stability and unfolding of cyt c were extensively studied using deuterium exchange [103] and other experimental techniques [104, 105 reviewed in ref. 97]. Stabilities of different regions of the protein were very dissimilar: five distinct domains of cyt c (foldons) with nonequivalent stabilities were identified, which participate in cooperative folding–unfolding of the protein in a stepwise sequential way [103]. These structural domains are folded around the heme of cyt c, which is covalently attached to the polypeptide chain by residues Cys14 and Cys17.
Accordingly, the new concepts in drug discovery based on the design of mitochondria-targeted inhibitors of cyt c/CL peroxidase and CL peroxidation with antiapoptotic effects have appeared. Therefore, mitochondria-targeted disruptors and inhibitors of cyt c/CL peroxidase complexes and suppression of CL peroxidation represent new strategies in anti-apoptotic drug discovery [106].
We have originally discovered that both carnosine and carcinine (10-25 mM) are capable of inhibiting the catalysis of linoleic acid and phosphatidylcholine liposomal peroxidation (LPO) by the O2ˉ˙-dependent iron-ascorbate and lipid-peroxyl-radical-generating linoleic acid 13-monohydroperoxide (LOOH)-activated haemoglobin systems, as measured by thiobarbituric-acid-reactive substance. Carcinine and carnosine are good scavengers of OH˙ radicals, as detected by iron-dependent radical damage to the sugar deoxyribose. This suggests that carnosine and carcinine are able to scavenge free radicals or donate hydrogen ions. The iodometric, conjugated diene and t.l.c. assessments of lipid hydroperoxides (13-monohydroperoxide linoleic acid and phosphatidylcholine hydroperoxide) showed their efficient reduction and deactivation by carnosine and carcinine (10-25 mM) in the liberated and bound-to-artificial-bilayer states. This suggests that the peroxidase activity exceeded that susceptible to direct reduction with glutathione peroxidase. Imidazole, solutions of beta-alanine, or their mixtures with peptide moieties did not show antioxidant potential [107]. Due to the combination of weak metal chelating (abolished by EDTA), OH˙ and lipid peroxyl radicals scavenging, reducing activities to liberated fatty acid and phospholipid hydroperoxides, carnosine and carcinine appear to be physiological antioxidants able to efficiently protect the lipid phase of biological membranes and aqueous environments and act as the anti-apoptotic natural drug compounds. Removal of excessive mitochondrial ROS by electron scavengers and antioxidants is a promising therapeutic strategy to reduce the detrimental effects of UV radiation and other cataractogenic factors exposure. Local mitochondrial interaction is mediated by superoxide (O2ˉ˙ ) diffusion and the O2ˉ˙ -dependent activation of an inner membrane anion channel (IMAC). In contrast to other mitochondria-targeted antioxidants studied by Mitchel and Skulachev’s Groups, it is important that L-carnosine can scavenge superoxide anion radical released from mitochondria in the outdside compartments of the cells. Even at low concentrations, dipeptide carnosine forms a charge-transfer complex (Car ... O2ˉ˙ , lambda max = 265 nm) with the superoxide radical which changes the reactivity of O2ˉ˙. The absorbance band of the complex was shifted towards lower energy as compared to superoxide radical lambda max = 255 nm). The interaction of carnosine with OH-radicals proceeding at very high rate and resulting in the formation of a stable product suggested another type of dipeptide activity [108].
The IVP patented concept includes the integrated lens and ocular tissues mitochondrial targeting with triphenyl-phosphonium (TPP)-conjugated hydrophobic compounds combined with L-carnosine ophthalmic prodrug N-acetylcarnosine (Figures 8, 9), represents a promising approach for the development of novel eye and specifically, cataract universal antioxidant protectors [89, 90]. N-acetylcarnosine eye-drops have been shown to have measurable effects to prevent and partially reverse cataracts within only 3-6-month of topical ocular use [92, 109]. However, it is recommended that for maximum efficacy, that administration be continued for a period not less than 3-5 months. In addition, the drops’ effectiveness is increased the sooner they are used after a cataract is detected. Also, considering that senile cataracts are an on-going aging disorder, N-acetylcarnosine may be required on a regular basis to help maintain the eye's natural anti-oxidant defenses. Other than senile cataract, N-acetylcarnosine may have other benefits. The unique N-acetylcarnosine formula with its added and synergistic lubricants, could also provide beneficial results with the following eye-disorders [92, 109]:
• Presbyopia.
• Open-angle primary glaucoma (in combination with beta-blockers).
• Corneal disorders.
• Computer vision syndrome.
• Eye strain.
• Ocular inflammation.
• Blurred vision.
• Dry eye syndrome.
• Retinal diseases.
• Vitreous opacities and lesions.
• Complications of diabetes mellitus and other systemic diseases.
• Contact lens difficulties, particularly with soft contact lenses. (Not only do the lubricants in the Can-C N-acetylcarnosine eye-drop help to make wearing contact lenses more comfortable, but n-acetylcarnosine is also believed to reduce the build up of lactic acid in the eye, thus enabling the lens to be left safely in the eye for longer).
Mitochondria are usually either the primary source of ROS [110] or mediators of burst of ROS formation in other cellular compartments (“ROS-induced ROS release” [12, 111]). This is why it is reasonable to combine the N-acetylcarnosine lubricant eye drops acting as the ophthalmic prodrug of naturally targeted to mitochondria L-carnosine endowed with pluropotent types of antioxidant activities with mitochondria-targeted rechargeable antioxidant (see a number of compounds presented in Figure 8) as a medicine to treat ocular diseases [reviewed in ref. [12]].
Altered redox and cytokine patterns suggest inhibition of expression/activity of metabolizing and antioxidant enzymes in tissues subjected to the action of oxidative stress. Metabolic parameters indicating accelerated LPO, increased nitric oxide production and glutathione depletion in combination with increased inflammatory cytokines should be considered in biological definition and diagnosis of ocular disorders. We suggest that pharmacologic loading of intact cells with lipophilic antioxidants may compromise the yield of valuable proprietary antioxidant enzymatic activities, such as superoxide dismutase and glutathione reductase activities and have the pathogenic importance of intracellular ROS in antioxidant overload states deprivation, inflammation, and cellular injury. The excessive treatment with lipophilic xenobiotic compound can lead to decrease of antioxidant enzyme activities, namely, superoxide dismutase (SOD), catalase (CAT), glutathione peroxidase (GPx) and glutathione reductase (GR) associated with a higher amount of MDA level. Although ROS are essential participants in cell signaling and regulation, when their cellular production overwhelms the intrinsic antioxidant capacity, damage to cellular macromolecules such as DNA, proteins, and lipids ensues. Oxidative stress contributes to the pathogenesis of a number of the ocular neurodegenerative diseases that might be protected with the described here with therapeutic modality. The combined pharmacological use of N-acetylcarnosine eye drop prodrug-codrug formulation as an ocular promoter of L-carnosine activated by a triphenylphosphonium cation coupled bioactive molecules delivered to mitochondria can prevent the impediment of antioxidant enzymatic activities and can prevent the exacerbation of ophthalmic sight threatening disease [12].
Attractive Targets and Therapeutic Strategies of Drug Delivery to Mitochondria in the Treatment of Oxidative Injury During Glaucomatous Neurodegeneration
Although retinal ganglion cells (RGCs) exhibit unique characteristics of antioxidant defense mechanisms [112], survival of axotomized RGCs is critically sensitive to the oxidative redox state; and decreasing ROS generation promotes the survival of RGCs after axotomy [113]. Retinal light exposure is a source of oxidative stress, and retinal cells contain molecules that scavenge or inactivate ROS. Yet, ROS also play a role in signal transduction, and some retinal cells (e.g., neurotrophin-dependent retinal ganglion cells, RGCs) may use ROS as part of the signaling process for cell death. RGCs might therefore have specialized mechanisms for regulating ROS levels. The hypothesis that RGCs might regulate ROS differently from other retinal cells was tested by studying their differential response to oxidative stress in vitro [112]. Growing evidence also supports that the oxidative stress evident in glaucomatous tissues is an important factor for the loss of these neurons during glaucomatous neurodegeneration. Most of the proposed pathogenic mechanisms of glaucomatous neurodegeneration are well-known to include an oxidative component. Histopathological studies of glaucomatous human donor eyes [114, 115] and experimental studies using different animal models of glaucoma have demonstrated that while optic nerve axons are progressively lost in glaucomatous eyes, RGCs die through apoptosis [11, 116-118]. The results suggest that retinal ganglion cells of the adult rat die through apoptosis when the IOP is markedly increased. This raises new possibilities in the treatment of glaucomatous damage to the retina, by the potential interruptibility of a program for neuronal death [116]. Multiple pathogenic mechanisms triggered by elevated IOP and/or tissue hypoxia have been associated with optic nerve degeneration and RGC apoptosis in glaucoma. A number of genetic susceptibility factors also support the multifactorial nature of the disease [119, 120]. During glaucomatous neurodegeneration, ROS may be directly neurotoxic to RGCs, may contribute to secondary degeneration by inducing glial dysfunction, and may also function as signaling molecules by acting as a second messenger and/or by regulating redox modifications of downstream effectors [11]. Exact molecular mechanisms and the real impact of oxidative stress on the development and progression of glaucomatous neurodegeneration need to be further elucidated. An improved understanding of the cellular mechanisms leading to oxidative injury in RGCs during glaucomatous neurodegeneration, and modulation of pathways involved in mediating cellular responses to oxidative stress, may offer unique opportunities for neuroprotective interventions aimed at the effective treatment of glaucoma, a leading cause of blindness [11].
In this work we proposed the combined use of mitochondria-targeted antioxidant mito Vit E and N-acetylcarnosine, an ophthalmic prodrug of L-carnosine in formulation of eye drops including the mucoadhesive compound carboxymethylcellulose to help elucidate the role of mitochondrial oxidative damage in apoptotic cell death (Figures 8,9) [12, 89, 90]. We suggest that mitochondrial oxidative damage plays an important role in ROS-induced apoptosis. Further work using these and other mitochondrially targeted compounds to dissect out the role of mitochondrial oxidative changes in peroxide-induced apoptosis is ongoing. The findings reported here demonstrate that mitochondrially targeted antioxidants such as mito vit E + N-acetylcarnosine in the eye drop formulation with carboxymethylcellulose can be used to investigate the role of mitochondrial oxidative stress in RGCs cell death. This strategy also has potential for unraveling the contribution of oxidative stress to other ocular pathologies involving mitochondrial dysfunction [12, 89, 90].
Conclusion
The mitochondrial respiratory chain is a major source of superoxide and consequently generated related ROS and, therefore, mitochondria accumulate oxidative damage more rapidly than the rest of the cell, contributing to mitochondrial dysfunction and cell death in ocular degenerative diseases and in aging [11, 12, 18, 89, 90]. Mitochondria are also central to activating apoptosis and oxidative damage can lead to cell death, however, the significance of mitochondrial oxidative damage for cell death is unclear [70, 121, 122]. The consequences of mitochondrial dysfunction (collapse of the mitochondrial inner transmembrane potential, uncoupling of the respiratory chain, hyperproduction of superoxide anions, disruption of mitochondrial biogenesis, outflow of matrix calcium and glutathione, and release of soluble intermembrane proteins) entails a bioenergetic catastrophe culminating in the disruption of plasma membrane integrity (necrosis) and/or the activation of specific apoptogenic proteases (caspases) by mitochondrial proteins that leak into the cytosol (cytochrome c, apoptosis- inducing factor) with secondary endonuclease activation (apoptosis). The relative rate of these two processes (bioenergetic catastrophe versus protease and endonuclease activation) determines whether a cell will undergo primary necrosis or apoptosis. The acquisition of the biochemical and ultrastructural features of apoptosis critically relies on the liberation of apoptogenic proteases or protease activators from mitochondria. The fact that mitochondrial events control cell death has major implications for the development of cytoprotective and cytotoxic drugs [122]. One approach to this problem is to selectively target antioxidants to mitochondria [12, 68-73].
This should allow the relative importance of mitochondrial and cytoplasmic oxidative stress for cell death to be distinguished, and also enable the contribution of mitochondrial damage to ocular pathologies associated with aging, diabetes, and apoptosis of retinal neuronal cells (RGCs) to be investigated in cell and animal models.
With the recognition of the central role of mitochondria in apoptosis, there is a need to develop specific tools to manipulate mitochondrial function within cells in ocular disorders, such as glaucoma, human cataract and retinal disorders. Here we report on the development of a novel antioxidant eye drop formulation N-acetylcarnosine eye drop prodrug–codrug as an ocular promoter of L-carnosine, a natural mitochondria-targeted antioxidant, activated by a triphenylphosphonium cation coupled bioactive molelcules delivered to mitochondria that selectively blocks mitochondrial oxidative damage, enabling the roles of mitochondrial oxidative stress in different types of ocular cell death to be inferred (Figure 7).
Due to the large mitochondrial membrane potential, the cation was accumulated within mitochondria inside cells, where the lipophilic moiety of mitochondria-targeted antioxidant inserted into the lipid bilayer and was reduced by the respiratory chain. Due to the combination of weak metal chelating (abolished by EDTA), OH˙ and lipid peroxyl radicals scavenging, reducing activities to liberated fatty acid and phospholipid hydroperoxides, carnosine derived in the ocular tissues and fluids from the ophthalmic prodrug N-acetylcarnosine, appears to be a physiological universal antioxidant able to efficiently protect the lipid phase of biological membranes and aqueous environments and act as the anti-apoptotic natural drug compound . After detoxifying a reactive oxygen species, the lipophilic moiety of mitochondria-targeted antioxidant was regenerated by the respiratory chain enabling its antioxidant activity to be recycled. We have shown that selectively manipulating mitochondrial antioxidant status with targeted dipeptide and recyclable antioxidants is a feasible approach to investigate the role of mitochondrial oxidative damage in apoptotic cell death in sight-threatening eye disorders. This approach will have further clinical applications in investigating mitochondrial dysfunction in a range of experimental models.
We conclude that, based on the data presented in this study, the combined eye drop formulation including N-acetylcarnosine prodrug–codrug as an ocular promoter of L-carnosine, a natural mitochondria-targeted antioxidant , activated by a triphenylphosphonium cation coupled bioactive molelcules delivered to mitochondria that selectively block mitochondrial oxidative damage, may represent new pharmacologic tool to mitigate complex ocular pathology for the treatment of sight-threatening eye diseases and, especially, neurodegeneration originating from an oxidative injury and glaucomatous neurodegeneration.
Acknowledgements
This work was planned, organized, and supported by Innovative Vision Products, Inc. (County of New Castle, DE, USA), by Russian Foundation for Basic Research (grant 09-04-01071a and Federal Agency for Education (State contract P1293) to Prof. Yegorov Y.E.) . Innovative Vision Products (IVP) is a holder of the worldwide Patents: (WO 2004/028536 A1; WO 94/19325; WO 95/12581; WO 2004/064866 A1) (including most relevant to the article issues PCT International Publication Number WO 2004/064866 PCT/JP2004/000351) protecting the therapeutic applications and formulations thereof of carnosine and carnosine derivatives stabilizing carnosine from enzymatic hydrolysis including tyhose with the entrapped mitochondria-targeted antioxidant moiety. Innovative Vision Products Inc. is a pharmaceutical and Nanotechnology Development Company with a focus on innovative chemical entities, drug delivery systems, and unique medical devices to target specific biomedical applications. Over the last decade IVP has developed a track record in developing these technologies to effectively address the unmet needs of specific diseased populations.
Conflict Of Interest
Declaration of interest: The author (Dr. Mark A. Babizhayev) reports the interest in the Intellectual Property of the described modalities protected with the patents. The authors bear primary responsibility for accuracy of made statements and employment of the described products and for the content and writing of the paper. Dr. Mark Babizhayev is the Senior Great Grandson of Wallis Simpson (Bessie Wallis Warfield-Spencer-Simpson) USA wife of King of UK Edward VIII “David” (Edward Albert Christian George Andrew Patrick David) Prince of Wales UK, Duke & Duchess of Windsor.
References
- Resnikoff S, Pascolini D, Etya'ale D, Kocur I, Pararajasegaram R, et al., (2004) Global data on visual impairment in the year 2002. Bull World Health Organ. 82(11): 844-51. 82(11): 844-51.
- Ohia SE, Opere CA, Leday AM (2005) Pharmacological consequences of oxidative stress in ocular tissues. Mutat Res. 579(1-2): 22-36.
- Augusteyn RC (1981) Protein modification in cataract: possible oxidative mechanisms. Mechanisms of Cataract Formation in the Human Lens Academic Press: (London). 71−116.
- Berman ER (1991) Biochemistry of the Eye. Plenum Press: New York.
- Justilien V, Pang JJ, Renganathan K, Zhan X, Crabb JW, et al., (2007) SOD2 knockdown mouse model of early AMD. Invest Ophthalmol Vis Sci. 48(10): 4407-20.
- King A, Gottlieb E, Brooks DG, Murphy MP, Dunaief JL (2004) Mitochondria-derived reactive oxygen species mediate blue light-induced death of retinal pigment epithelial cells. Photochem Photobiol. 79(5): 470-5.
- Komeima K, Rogers BS, Lu L, Campochiaro PA (2006) Antioxidants reduce cone cell death in a model of retinitis pigmentosa. Proc Natl Acad Sci U S A. 103(30) : 11300-5.
- Komeima K, Rogers BS, Campochiaro PA (2007) Antioxidants slow photoreceptor cell death in mouse models of retinitis pigmentosa. J Cell Physiol. 213(3): 809-15.
- Ghelli A, Porcelli AM, Zanna C, Martinuzzi A, Carelli V, et al., (2008) Protection against oxidant-induced apoptosis by exogenous glutathione in Leber hereditary optic neuropathy cybrids. Invest Ophthalmol Vis Sci. 49(2): 671-6.
- McKinnon SJ (2003) Glaucoma: ocular Alzheimer's disease? Front Biosci. 8: s1140-56.
- Tezel G (2006) Oxidative stress in glaucomatous neurodegeneration: mechanisms and consequences. Prog Retin Eye Res. 25(5): 490-513.
- Babizhayev MA, Yegorov YE (2010) Reactive Oxygen Species and the Aging Eye: Specific Role of Metabolically Active Mitochondria in Maintaining Lens Function and in the Initiation of the Oxidation-Induced Maturity Onset Cataract-A Novel Platform of Mitochondria-Targeted Antioxidants With Broad Therapeutic Potential for Redox Regulation and Detoxification of Oxidants in Eye Diseases. Am J Ther. 23(1): e98-117.
- Babizhayev MA, Costa EB (1994) Lipid peroxide and reactive oxygen species generating systems of the crystalline lens. Biochim Biophys Acta. 1225(3): 326-37.
- Olofsson EM, Marklund SL, Behndig A (2007) Glucose-induced cataract in CuZn-SOD null lenses: an effect of nitric oxide? Free Radic Biol Med. 42(7): 1098-105.
- Lassen N, Bateman JB, Estey T, Kuszak JR, Nees DW, et al., (2007) Multiple and additive functions of ALDH3A1 and ALDH1A1: cataract phenotype and ocular oxidative damage in Aldh3a1(-/-)/Aldh1a1(-/-) knock-out mice. J Biol Chem. 282(35): 25668-76.
- Brito BE, Marcano JC, Salazar E, Cano M, Baute L, et al., (2006) Age as a determinant factor for endotoxin induced uveitis. Ocul Immunol Inflamm. 14(2): 117-24.
- Pararajasegaram G, Sevanian A, Rao NA (1991) Suppression of S antigeninduced uveitis by vitamin E supplementation. Ophthalmic Res. 23(3): 121-7.
- Antonenko YN, Avetisyan AV, Bakeeva LE, Chernyak BV, Chertkov VA, et al., (2008) Mitochondria-targeted plastoquinone derivatives as tools to interrupt execution of the aging program. 1. Cationic plastoquinone derivatives: synthesis and in vitro studies. Biochemistry (Mosc). 73(12): 1273-87.
- Quigley HA (1996) Number of people with glaucoma worldwide. Br J Ophthalmol. 80(5): 389-93.
- Webb M (1987) Toxicological significance of metallothionein. Exp Suppl. 52: 109−134.
- Fletcher AE (2010) Free radicals, antioxidants and eye diseases: evidence from epidemiological studies on cataract and age-related macular degeneration. Ophthalmic Res. 44(3): 191-8.
- Brennan LA, Kantorow M (2009) Mitochondrial function and redox control in the aging eye: role of MsrA and other repair systems in cataract and macular degenerations. Exp Eye Res. 88(2): 195-203.
- Babizhayev MA, Yegorov YE (2010) Smoking and health: association between telomere length and factors impacting on human disease, quality of life and life span in a large population-based cohort under the effect of smoking duration. Fundam Clin Pharmacol. 25(4): 425-42.
- Babizhayev MA, Savel’yeva EL, Moskvina SN, Yegorov YE (2010) Telomere Length is a Biomarker of Cumulative Oxidative Stress, Biologic Age, and an Independent Predictor of Survival and Therapeutic Treatment Requirement Associated With Smoking Behavior. Am J Ther. 18(6): e209-26.
- Zarbin MA, Rosenfeld PJ (2010) Pathway-based therapies for age-related macular degeneration: an integrated survey of emerging treatment alternatives. Retina. 30(9): 1350-67.
- Zarbin M, Szirth B (2007) Current treatment of age-related macular degeneration. Optom Vis Sci. 84(7): 559-72.
- Jarrett SG, Lewin AS, Boulton ME (2010) The importance of mitochondria in age-related and inherited eye disorders. Ophthalmic Res. 44(3): 179-90.
- Bantseev V, Youn HY (2006) Mitochondrial "movement" and lens optics following oxidative stress from UV-B irradiation: cultured bovine lenses and human retinal pigment epithelial cells (ARPE-19) as examples. Ann N Y Acad Sci. 1091: 17-33.
- Yao K, Zhang L, Zhang Y, Ye P, Zhu N (2008) The flavonoid, fisetin, inhibits UV radiation-induced oxidative stress and the activation of NF-kappaB and MAPK signaling in human lens epithelial cells. Mol Vis. 14: 1865-71.
- Yao K, Ye P, Zhang L, Tan J, Tang X, et al., (2008) Epigallocatechin gallate protects against oxidative stress-induced mitochondria-dependent apoptosis in human lens epithelial cells. Mol Vis. 14: 217-23.
- Ottonello S, Foroni C, Carta A, Petrucco S, Maraini G (2000) Oxidative stress and age-related cataract. Ophthalmologica. 214(1): 78-85.
- Spector A (1995) Oxidative stress-induced cataract: mechanism of action. FASEB J. 9(12): 1173-82.
- Truscott RJ (2005) Age-related nuclear cataract-oxidation is the key. Exp Eye Res. 80(5): 709-25.
- Huang L, Estrada R, Yappert MC, Borchman D (2006) Oxidation-induced changes in human lens epithelial cells. 1. Phospholipids. Free Radic Biol Med. 41(9): 1425-32.
- Yan Q, Liu JP, Li DW (2006) Apoptosis in lens development and pathology. Differentiation. 74(5): 195-211.
- Li WC, Kuszak JR, Dunn K, Wang RR, Ma W, et al., (1995) Lens epithelial cell apoptosis appears to be a common cellular basis for non-congenital cataract development in humans and animals. J Cell Biol. 130(1): 169-81.
- Yao K, Tan J, Gu WZ, Ye PP, Wang KJ (2007) Reactive oxygen species mediates the apoptosis induced by transforming growth factor beta(2) in human lens epithelial cells. Biochem Biophys Res Commun. 354(1): 278-83.
- Kilic F, Trevithick JR (1998) Modelling cortical cataractogenesis. XXIX. Calpain proteolysis of lens fodrin in cataract. Biochem Mol Biol Int. 45(5): 963-78.
- Kilic F, Handelman GJ, Traber K, Tsang K, Packer L, et al., (1998) Model ling cortical cataractogenesis XX. in vitro effect of alpha-lipoic acid on glutathione concentrations in lens in model diabetic cataractogenesis. Biochem Mol Biol Int. 46(3): 585-95.
- Kilic F, Handelman GJ, Serbinova E, Packer L, Trevithick JR (1995) Modelling cortical cataractogenesis 17: in vitro effect of a-lipoic acid on glucoseinduced lens membrane damage, a model of diabetic cataractogenesis. Biochem Mol Biol Int. 37(2): 361-70.
- Bantseev VL, Herbert KL, Trevithick JR, Sivak JG (1999) Mitochondria of rat lenses: distribution near and at the sutures. Curr Eye Res. 19(6): 506-16.
- Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, et al., (1997) Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 91(4): 479-89.
- Rattner A, Nathans J (2006) Macular degeneration: recent advances and therapeutic opportunities. Nat Rev Neurosci. 7(11): 860-72.
- Khandhadia S, Lotery A (2010) Oxidation and age-related macular degeneration: insights from molecular biology. Expert Rev Mol Med. 12: e34.
- Osborne NN, Kamalden TA, Majid AS, Del Olmo-Aguado S, Manso AG, et al., (2010) Light Effects on Mitochondrial Photosensitizers in Relation to Retinal Degeneration. Neurochem Res.35(12): 2027-34.
- Jarrett SG, Lin H, Godley BF, Boulton ME (2008) Mitochondrial DNA damage and its potential role in retinal degeneration. Prog Retin Eye Res. 27(6): 596-607.
- Pedersen PL (1999) Mitochondrial events in the life and death of animal cells: a brief overview. J Bioenerg Biomembr. 31(4): 291-304.
- Harper ME, Bevilacqua L, Hagopian K, Weindruch R, Ramsey JJ (2004) Ageing, oxidative stress, and mitochondrial uncoupling. Acta Physiol Scand. 182(4): 321-31.
- Orrenius S (2007) Reactive oxygen species in mitochondria-mediated cell death. Drug Metab Rev. 39(2-3): 443-55.
- Wallace DC (1999) Mitochondrial diseases in man and mouse. Science. 283(5407): 1482-8.
- Ott M, Gogvadze V, Orrenius S, Zhivotovsky B (2007) Mitochondria, oxidative stress and cell death. Apoptosis. 12(5): 913-22.
- Hudson EK, Hogue BA, Souza-Pinto NC, Croteau DL, Anson RM, et al., (1998) Age-associated change in mitochondrial DNA damage. Free Radic Res. 29(6): 573-9.
- Caron F, Jacq C, Rouvière-Yaniv J (1979) Characterization of a histonelike protein extracted from yeast mitochondria. Proc Natl Acad Sci U S A. 76(9): 4265-9.
- Beckman KB, Ames BN (1998) The free radical theory of aging matures. Physiol Rev. 78(2): 547-81.
- Shigenaga MK, Hagen TM, Ames BN (1994) Oxidative damage and mitochondrial decay in aging. Proc Natl Acad Sci U S A. 91(23): 10771-8.
- Green D, Kroemer G (1998) The central executioners of apoptosis: caspases or mitochondria? Trends Cell Biol. 8(7): 267-71.
- Duke RC, Ojcius DM, Young JD (1996) Cell suicide in health and disease. Sci Am. 275(6): 80-7.
- Kong GY, Van Bergen NJ, Trounce IA, Crowston JG (2009) Mitochondrial dysfunction and glaucoma. J Glaucoma. 18(2): 93-100.
- Chrysostomou V, Trounce IA, Crowston JG (2010) Mechanisms of retinal ganglion cell injury in aging and glaucoma. Ophthalmic Res. 44(3): 173-8.
- Munemasa Y, Kitaoka Y, Kuribayashi J, Ueno S (2010) Modulation of mitochondria in the axon and soma of retinal ganglion cells in a rat glaucoma model. J Neurochem. 115(6): 1508-19.
- Dhanasekaran A, Kotamraju S, Kalivendi SV, Matsunaga T, Shang T, et al., (2004) Supplementation of endothelial cells with mitochondria-targeted antioxidants inhibit peroxide-induced mitochondrial iron uptake, oxidative damage, and apoptosis. J Biol Chem. 279(36): 37575-87.
- Michikawa Y, Mazzucchelli F, Bresolin N, Scarlato G, Attardi G (1999) Aging-dependent large accumulation of point mutations in the human mtDNA control region for replication. Science. 286(5440): 774-9.
- Bharath S, Hsu M, Kaur D, Rajagopalan S, Andersen JK (2002) Glutathione, iron and Parkinson's disease. Biochem Pharmacol. 64(5-6): 1037- 48.
- Gu M, Owen AD, Toffa SE, Cooper JM, Dexter DT, et al., (1998) Mitochondrial function, GSH and iron in neurodegeneration and Lewy body diseases. J Neurol Sci. 158(1): 24-9.
- Kasarskis EJ, Tandon L, Lovell MA, Ehmann WD (1995) Aluminum, calcium, and iron in the spinal cord of patients with sporadic amyotrophic lateral sclerosis using laser microprobe mass spectroscopy: a preliminary study. J Neurol Sci. 130(2): 203-8.
- Bartzokis G, Cummings J, Perlman S, Hance DB, Mintz J (1999) Increased basal ganglia iron levels in Huntington disease. Arch Neurol. 56(5): 569-74.
- Dexter DT, Carayon A, Javoy-Agid F, Agid Y, Wells FR, et al., (1991) Alterations in the levels of iron, ferritin and other trace metals in Parkinson's disease and other neurodegenerative diseases affecting the basal ganglia. Brain. 114(Pt 4): 1953-75.
- Murphy MP, Smith RA (2000) Drug delivery to mitochondria: the key to mitochondrial medicine. Adv Drug Deliv Rev. 41(2): 235-50.
- Murphy MP (1997) Selective targeting of bioactive compounds to mitochondria. Trends Biotechnol. 15(8): 326-30.
- Kelso GF, Porteous CM, Coulter CV, Hughes G, Porteous WK, et al., (2000) Selective targeting of a redox-active ubiquinone to mitochondria within cells: antioxidant and antiapoptotic properties. J Biol Chem. 276(7): 4588-96.
- Liberman EA, Topaly VP, Tsofina LM, Jasaitis AA, Skulachev VP (1969) Mechanism of coupling of oxidative phosphorylation and the membrane potential of mitochondria. Nature. 222(5198): 1076-8.
- Murphy MP, Smith RA (2007) Targeting antioxidants to mitochondria by conjugation to lipophilic cations. Annu Rev Pharmacol Toxicol. 47: 629– 656.
- Smith RA, Kelso GF, James AM, Murphy MP (2004) Targeting coenzyme Q derivatives to mitochondria. Methods Enzymol. 382: 45–67.
- Matthews RT, Yang L, Browne S, Baik M, Beal MF (1998) Coenzyme Q10 administration increases brain mitochondrial concentrations and exerts neuroprotective effects. Proc Natl Acad Sci U S A. 95(15): 8892-7.
- Lass A, Sohal RS (1998) Electron transport-linked ubiquinone-dependent recycling of alpha-tocopherol inhibits autooxidation of mitochondrial membranes. Arch Biochem Biophys. 352(2): 229-36.
- Crane FL (1977) Hydroquinone dehydrogenases. Annu Rev Biochem. 46: 439-69.
- Maguire JJ, Wilson DS, Packer L (1989) Mitochondrial electron transportlinked tocopheroxyl radical reduction. J Biol Chem. 264(36): 21462-5.
- [78]. Kagan VE, Serbinova EA, Stoyanovsky DA, Khwaja S, Packer L (1994) Assay of ubiquinones and ubiquinols as antioxidants. Methods Enzymol. 234: 343-54.
- Ernster L, Forsmark P, Nordenbrand K (1992) The mode of action of lipidsoluble antioxidants in biological membranes: relationship between the effects of ubiquinol and vitamin E as inhibitors of lipid peroxidation in submitochondrial particles. J Biofactors. 3(4): 241-8.
- Ingold KU, Bowry VW, Stocker R, Walling C (1993) Autoxidation of lipids and antioxidation by alpha-tocopherol and ubiquinol in homogeneous solution and in aqueous dispersions of lipids: unrecognized consequences of lipid particle size as exemplified by oxidation of human low density lipoprotein. Proc Natl Acad Sci U S A. 90(1): 45-9.
- Land EJ, Swallow AJ (1970) One-electron reactions in biochemical systems as studied by pulse radiolysis. 3. Ubiquinone. J Biol Chem. 245(8): 1890-4.
- Mukai K, Kikuchi S, Urano S (1990) Stopped-flow kinetic study of the regeneration reaction of tocopheroxyl radical by reduced ubiquinone-10 in solution. Biochim Biophys Acta. 1035(1): 77-82.
- Skulachev VP (2007) A biochemical approach to the problem of aging: "megaproject" on membrane-penetrating ions. The first results and prospects. Biochemistry (Mosc). 72(12): 1385-96.
- Skulachev VP, Antonenko YN, Cherepanov DA, Chernyak BV, Izyumov DS, et al., (2010) Prevention of cardiolipin oxidation and fatty acid cycling as two antioxidant mechanisms of cationic derivatives of plastoquinone (SkQs). Biochim Biophys Acta. 1797(6-7): 878-89.
- Loshadkin D, Roginsky V, Pliss E (2002) Substituted p-hydroquinones as a chain-breaking antioxidant during the oxidation of styrene. Int J Chem Kinetics. 34(3): 162-171.
- Roginsky V, Barsukova T, Loshadkin D, Pliss E (2003) Substituted p-hydroquinones as inhibitors of lipid peroxidation. Chem Phys Lipids. 125(1): 49-58.
- Kruk J, Jemiola-Rzeminska M, Strzalka K (1997) Plastoquinol and α-tocopherol quinol are more active than ubiquinol and α-tocopherol in inhibition of lipid peroxidation. Chem Phys Lipids. 87(1): 73-80.
- Huang L, Tang D, Yappert MC, Borchman D (2006) Oxidation-induced changes in human lens epithelial cells 2. Mitochondria and the generation of reactive oxygen species. Free Radic Biol Med. 41(6): 926-36.
- Babizhayev MA, Meguro K (2004) Combined use of carnosinase inhibitor with l-carnosines and composition. WO 2004/064866 PCT/ JP2004/000351.
- Babizhayev MA (2004) Method for topical treatment of eye disease and composition and device for said treatment. PCT Patent Application. International Publication Number WO 2004/028536 A1.
- Babizhayev MA (2006) Ophthalmic pharmacology of N-acetylcarnosine lubricant eye drops . J Pharmacology Toxicology 1(3): 201-233.
- Babizhayev MA, Micans P, Guiotto A, Kasus-Jacobi A (2009) N-acetylcarnosine lubricant eyedrops possess all-in-one universal antioxidant protective effects of L-carnosine in aqueous and lipid membrane environments, aldehyde scavenging, and transglycation activities inherent to cataracts: a clinical study of the new vision-saving drug N-acetylcarnosine eyedrop therapy in a database population of over 50,500 patients. Am J Ther. 16(6): 517-33.
- Zakharchenko MV, Temnov AV, Kondrashova MN (2003) Effect of carnosine on self-organization of mitochondrial assemblies in rat liver homogenate. Biochemistry (Mosc). 68(9): 1002-5.
- Kang KS, Yun JW, Lee YS (2002) Protective effect of L-carnosine against 12-O-tetradecanoylphorbol-13-acetate- or hydrogen peroxide-induced apoptosis on v-myc transformed rat liver epithelial cells. Cancer Lett. 178(1): 53-62.
- Korobov VN, Doliba NM, Telegus IaV (1993) [Carnosine in adaptation to hypobaric hypoxia]. Biokhimiia. 58(5): 740-4.
- Belikova NA, Tyurina YY, Borisenko G, Tyurin V, Samhan Arias AK, et al., (2009) Heterolytic reduction of fatty acid hydroperoxides by cytochrome c/ cardiolipin complexes: antioxidant function in mitochondria. J Am Chem Soc. 131(32): 11288-9.
- Kagan VE, Bayir HA, Belikova NA, Kapralov O, Tyurina YY, et al., (2009) Cytochrome c/cardiolipin relations in mitochondria: a kiss of death. Free Radic Biol Med. 46(11): 1439-53. https://www.ncbi.nlm.nih.gov/pubmed/19285551
- Tyurin VA, Tyurina YY, Jung MY, Tungekar MA, Wasserloos KJ, et al., (2009) Mass-spectrometric analysis of hydroperoxy- and hydroxy-derivatives of cardiolipin and phosphatidylserine in cells and tissues induced by pro-apoptotic and pro-inflammatory stimuli. J Chromatogr B Analyt Technol Biomed Life Sci. 877(26): 2863-72.
- Pinheiro TJ, Watts A (1994) Lipid specificity in the interaction of cytochrome c with anionic phospholipid bilayers revealed by solid-state 31P NMR. Biochemistry. 33(9): 2451-8.
- Pinheiro TJ, Elöve GA, Watts A, Roder H (1997) Structural and kinetic description of cytochrome c unfolding induced by the interaction with lipid vesicles. Biochemistry. 36(42): 13122-32.
- Muga A, Mantsch HH, Surewicz WK (1991) Membrane binding induces destabilization of cytochrome c structure. Biochemistry. 30(29): 7219-24.
- Spooner PJ, Watts A (1992) Cytochrome c interactions with cardiolipin in bilayers: a multinuclear magic-angle spinning NMR study. Biochemistry. 31(41): 10129-38.
- Maity H, Maity M, Englander SW (2004) How cytochrome c folds, and why: submolecular foldon units and their stepwise sequential stabilization. J Mol Biol. 343(1): 223-33.
- Berners-Price SJ, Bertini I, Gray HB, Spyroulias GA, Turano P (2004) The stability of the cytochrome c scaffold as revealed by NMR spectroscopy. J Inorg Biochem. 98(5): 814-23.
- Naeem A, Khan RH (2004) Characterization of molten globule state of cytochrome c at alkaline, native and acidic pH induced by butanol and SDS. Int J Biochem Cell Biol. 36(11): 2281-92.
- Kagan VE, Bayir A, Bayir H, Stoyanovsky D, Borisenko GG, et al., (2009) Mitochondria-targeted disruptors and inhibitors of cytochrome c/cardiolipin peroxidase complexes: a new strategy in anti-apoptotic drug discovery.Mol Nutr Food Res. 53(1): 104-14.
- Babizhayev MA, Seguin M-C, Gueyne J, Evstigneeva RP, Ageyeva EA, et al., (1994) L-Carnosine (β-alanyl-L-histidine) and carcinine (β -alanylhistamine) act as natural antioxidants with hydroxyl-radical-scavenging and lipid peroxidase activities. Biochem J. 304(2): 509-16.
- Pavlov AR, Revina AA, Dupin AM, Boldyrev AA, Yaropolov AI (1993) The mechanism of interaction of carnosine with superoxide radicals in water solutions. Biochim Biophys Acta. 1157(3): 304-12.
- Babizhayev MA, Kasus-Jacobi A (2009) State of the art clinical efficacy and safety evaluation of N-acetylcarnosine dipeptide ophthalmic prodrug. Principles for the delivery, self-bioactivation, molecular targets and interaction with a highly evolved histidyl-hydrazide structure in the treatment and therapeutic management of a group of sight-threatening eye diseases. Curr Clin Pharmacol. 4(1): 4-37.
- Skulachev VP, Longo VD (2005) Aging as a mitochondria-mediated atavistic program: can aging be switched off? Ann N Y Acad Sci. 1057: 145-64.
- Zorov DB, Filburn CR, Klotz LO, Zweier JL, Sollott SJ (2000) Reactive oxygen species (ROS)-induced ROS release: a new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J Exp Med. 192(7): 1001-14.
- Kortuem K, Geiger LK, Levin LA (2000) Differential susceptibility of retinal ganglion cells to reactive oxygen species. Invest Ophthalmol Vis Sci. 41(10): 3176-82.
- Geiger LK, Kortuem KR, Alexejun C, Levin LA (2002) Reduced redox state allows prolonged survival of axotomized neonatal retinal ganglion cells. Neuroscience. 109(3): 635-42.
- Kerrigan LA, Zack DJ, Quigley HA, Smith SD, Pease ME (1997) TUNELpositive ganglion cells in human primary open-angle glaucoma. Arch Ophthalmol. 115(8): 1031-5.
- Wax MB, Tezel G, Edward PD (1998) Clinical and ocular histopathological findings in a patient with normal-pressure glaucoma. Arch Ophthalmol. 116(8): 993-1001.
- Garcia-Valenzuela E, Shareef S, Walsh J, Sharma SC (1995) Programmed cell death of retinal ganglion cells during experimental glaucoma. Exp Eye Res. 61(1): 33-44.
- Li Y, Schlamp CL, Nickells RW (1999) Experimental induction of retinal ganglion cell death in adult mice. Invest Ophthalmol Vis Sci. 40(5): 1004-8.
- Quigley HA, Nickells RW, Kerrigan LA, Pease ME, Thibault DJ, et al., (1995) Retinal ganglion cell death in experimental glaucoma and after axotomy occurs by apoptosis. Invest Ophthalmol Vis Sci. 36(5): 774-86.
- Libby RT, Gould DB, Anderson MG, John SW (2005) Complex genetics of glaucoma susceptibility. Annu Rev Genomics Hum Genet. 6:15-44.
- Sarfarazi M, Rezaie T (2003) Optineurin in primary open angle glaucoma. Ophthalmol Clin North Am. 16(4): 529-41.
- Polyak K, Xia Y, Zweier JL, Kinzler KW, Vogelstein B (1997) A model for p53-induced apoptosis. Nature. 389(6648): 300-5.
- Kroemer G, Dallaporta B, Resche-Rigon M (1998) The mitochondrial death/life regulator in apoptosis and necrosis. Annu Rev Physiol. 60: 619- 42.





