Compliment Receptor Type - 1 (CD35) Gene Polymorphism and Plasmodium falciperum Malaria
Saket RD*, Kol S, Tripathi AK, Singh SP, Tipathi J, Chauhan UK
Centre for Biotechnology studies, A.P.S. University, Rewa (M.P.), India.
*Corresponding Author
Rishabh Dev Saket,
Centre for Biotechnology studies,
A.P.S. University, Rewa (M.P.), India.
E-mail: rdev47@gmail.com
Article Type: Research Article
Recieved: March 07, 2015; Accepted: April 14, 2015; Published: April 17, 2015
Citation: Saket RD et al., (2015) Compliment Receptor Type - 1 (CD35) Gene Polymorphism and Plasmodium Falciperum Malaria. Int J Med Biotechnol Genetics. 03(3), 27-33. doi: dx.doi.org/10.19070/2379-1020-150005
Copyright: Saket RD© 2015. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
Abstract
Malaria is a most causative agent for worldwide death. Plasmodium falciperum infected malaria most dangerous than other plasmodium species. It has closely association to compliment receptor type-1 (CD35) gene polymorphism. CD35 (CR1) is a cell surface receptor for plasmodium falciperum containing PfEMP-1 as a legend. Density of CD35 on erythrocyte can be determined by CR1 allele (HH, HL, and LL). HH allele of CR1 gene express high density of CD35 whereas LL in low density. High density of CD35 is more susceptible to falciperum infection. CD35 is also responsible for sever malaria. During plasmodium infection, pro-inflammatory cytokine like TNF-α, IFN-γ levels are increased. The elevated ratio of TNF-α/ IL10 indicates falciperum infection. Cell adhesion protein like VCAM, ICAM also mediate the malarial infection.
2.Introduction
3.Prevalence and Epidemiology of p. falciparum
3.1.Life Cycle of Plasmodium Falciparum Malaria
3.2.Pathophysiology of Plasmodium Falciparum Malaria
3.3.Complement Receptor Type - 1 (CD35)
3.4.Cytokines and Malarial Infection
4.Conclusion
5.References
Keywords
Plasmodium Falciperum; CD35; CR1 Allele; TNF-α; IL10; ICAM.
Introduction
Malaria is the most infectious and dangerous disease in the world. The World Health Organization (WHO) estimated 225 million malaria cases worldwide with 781,000 deaths due to Plasmodium infection per year. Four types of Plasmodium species (P. falciparum, P. vivax, P. malariae, and P. ovale) are responsible for almost all human infections. Plasmodium falciparum malaria is responsible for more than one million deaths that occur each year from malaria infection in Africa. Most of these deaths occur as a result of complications such as severe malaria associated anaemia (SMA) and cerebral malaria (CM) or coma [3]. Compliment Receptor 1 (CR1), a protein on RBC cells that having role in immune complex clearance. It’s also known as C3b/C4b receptor or CD35. In humans this protein is encoded by CR1 gene is located at on the long arm of chromosome 1 at band 32 (1q32) and lies within a complex of immunoregulatory genes. The Compliment Receptor 1 (CR1) gene polymorphism conform density of CD35 on RBC cells. The human CR1 binds to a major malarial adhesin, the P. falciparum erythrocyte membrane protein-one (PfEMP-1). High density of CR1 on erythrocyte indicates high risk of falciparum infection [1, 30, 48, 8].
Prevalence and Epidemiology of p. falciparum
Malaria affects the 300-500 million people each year in which 1-3 million people leading cause of death worldwide annually. There are five Plasmodium species that infect humans; Plasmodium falciparum, Plasmodium vivax, Plasmodium ovale, Plasmodium malariae and Plasmodium knowlesi. These species differ in their morphology, immunology, and geographic distribution. Among the five species that cause malaria in humans, Plasmodium falciparum (P. falciparum) is the most virulent resulting in the greatest number of complications and the great majority of malaria-related deaths in children under the age of five. The evolutionary history and geographical distribution of P. falciparum reflects a three-way interaction between the parasite, the host, and the Anopheles sp. mosquito (the vector for transmission). Circa 1900, prior to the widespread use of anti-malarials, the distribution of malaria reached the geographic latitudes of 64º north and 32º south [45, 17].
However, genetic history and the co-evolution of P. falciparum with humans suggest this has not always been the geographic model. The closest relative to the modern day P. falciparum is the chimpanzee malaria parasite, Plasmodium reichenowi. It has been argued that P. falciparum is of African origin because P. reichenowi is a parasite that infects African chimpanzees. Despite some controversy, it is generally accepted that the divergence of these two species of malaria occurred approximately 9-10 million years ago, prior to the divergence of humans from non-human primate relatives such as the chimpanzees. It is believed that the major spread of P. falciparum in Africa occurred during the “Agrarian Revolution” (4000-5000 years ago) when small nomadic groups began to establish larger settled communities; this lifestyle change provided ideal conditions for sustained P. falciparum transmission [18, 4] (Figure 1).
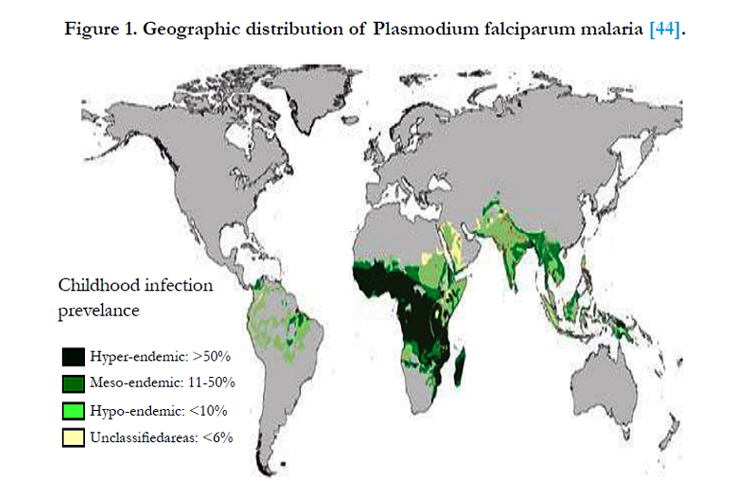
Figure 1. Geographic distribution of Plasmodium falciparum malaria [44].
The P. falciparum infection begins when a human host is bitten by an infected female Anopheles mosquito, and the mosquito injects sporozoites into the subcutaneous tissue of the human host. The sporozoites, within one hour reach to the liver and infect hepatocytes. The duration of the asymptomatic liver (exo-erythrocytic cycle) stage of the infection is approximately one-two weeks. During this stage, each sporozoite may yield thousands of merozoites [14].
The hepatocytes rupture releasing the merozoites into the blood stream (the beginning of clinical disease) where they are able to enter into RBCs by a complex invasion process comprised of four phases:
(a) initial recognition and reversible attachment of the merozoite to the RBC membrane.
(b) reorientation.
(c) invagination of the RBC membrane around the merozoite.
(d) resealing of the RBC membrane after completion of merozoite invasion.
RBC invasion is a rapid process that is governed by molecular interactions between the merozoites and the host cell surface [2,31].
Primary contact is initiated by a surface coat of proteins that is largely comprised of glycosylphosphatidylinositol (GPI)-anchored membrane proteins. There are at least nine recognized GPI anchored proteins that are predicted to be potential RBC ligands. Merozoite surface protein-1 (MSP-1) is the dominant antigen and is essential for parasite survival. MSP-1 is involved in the initial recognition of the RBC via sialic acid residues found on the RBC membrane. Other important proteins are MSP-2, -3 and -4. P. falciparum apical membrane antigen-1 (PfAMA-1) is also essential for successful invasion as it is translocated to the merozoites surface before invasion of the RBCs, and is also present on the sporozoite for invasion into hepatocytes [43, 2] (Figure 2).
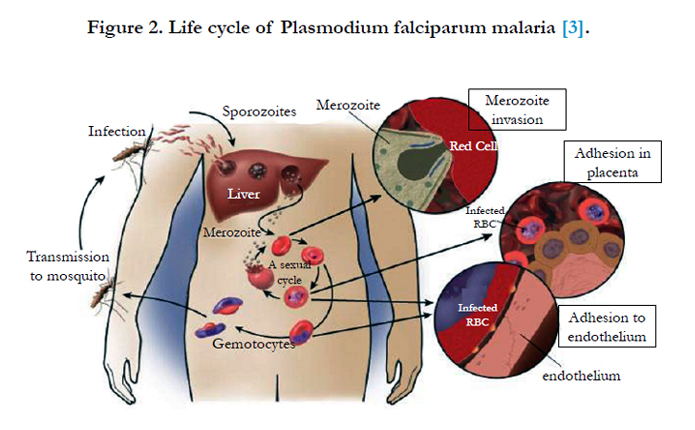
Figure 2. Life cycle of Plasmodium falciparum malaria [3].
Initially, the merozoites develop into an early trophozoite stage known as the “ring form”. The ring form persists for 24 hours and matures inside the RBC through a highly active metabolic state. The P. falciparum ring feeds from the host cytoplasm, importing glucose and breaking down hemoglobin into constituent amino acids. Following the ring stage, P. falciparum matures and develops to a late stage trophozite. The mature trophozoite stage parasite replicates by nuclear division resulting in schizont stage parasites. Each schizont is comprised of 20-24 merozoites, which are released upon rupture of the infected RBC. When the infected RBCs rupture, merozoites and parasite metabolic waste products such as hemozoin, degradation of hemoglobin, and parasite toxins are released. The majority of the merozoites will invade other RBCs continuing the asexual cycle; however, some parasites will form sexual stage forms called gametocytes which are then transmitted to new hosts by the Anopheles vector [31, 2, 5, 43, 45, 11, 32].
Infection with P. falciparum results in considerable morbidity and without treatment may be fatal. The clinical outcome of malaria depends on many contributing factors including the parasite’s virulence, the host’s response, geographical, and socio-economic factors (Table 1) [31]. The combination of these factors result in a range of possible outcomes for the host, including asymptomatic infection, uncomplicated malaria infection, severe infection (severe malaria anemia and cerebral malaria) and death.


Table 1. Factors contributing to the clinical outcome of P. falciparum infection [Adapted from [31]].
There are three defined clinical stages of malaria pathogenesis: uncomplicated malaria, severe malaria, and cerebral malaria. Uncomplicated malaria initially presents with fever and chills, nausea and headache, sometimes associated with diarrhea and vomiting. Unfortunately, because of the similarity in symptoms, malarial infection is often mistaken for many other infections including influenza or gastrointestinal infection and is therefore not properly treated [43]. In 1990, the World Health Organization (WHO) established criteria for the diagnosis of severe malaria. The major criteria include neurological involvement (cerebral malaria), pulmonary edema, acute renal failure, and severe anemia. Severe anemia is the second most common symptom of P. falciparum infection and is caused by the destruction of RBCs and overall decreased erythropoiesis. Acidosis and hypoglycemia are the most common metabolic complications [48, 51].
Cerebral malaria is the most common cause of death in adults and children with severe malaria. According to the WHO, the strict definition of cerebral malaria requires the presence of P. falciparum parasitemia and unarousable coma with a Glasgow Coma score of 9 or less; all other causes of coma, such as hypoglycemia, bacterial meningitis and viral encephalitis, need to be excluded. Typical neurological symptoms include coma, seizures, edema, and brainstem damage. Engorgement of cerebral capillaries and venules filled with infected RBCs and non-infected RBCs are typical histopathological findings in cerebral malaria. As the infection progresses, the increasingly detrimental pathogenesis of P. falciparum malaria is believed to be caused by two main factors: (a) an imbalance of cytokine production; and (b) the sequestration of infected RBCs in the microvasculature of vital organs [37, 19].
P. falciparum infection results in an increase of both pro-inflammatory cytokines and anti-inflammatory cytokines. However, in cerebral malaria, there is an unbalanced and excessive production of the pro-inflammatory response. Blood concentrations of pro-inflammatory cytokines, especially tumor necrosis factor (TNF), interferon gamma (IFN-γ), and IL- 6, have been shown to be raised in cerebral malaria. TNF may contribute to malaria pathogenesis including cerebral malaria. TNF up regulates endothelial cytoadherence receptors such as intercellular adhesion molecule-1 (ICAM-1), vascular cell adhesion molecule-1 (VCAM-1), and E-selectin. TNF may cause hypoglycemia and dyserthryopoesis, and has been shown to induce the release of nitric oxide (NO) which interferes with synaptic transmission [10, 21].
P. falciparum has a unique ability to adhere to host micro vasculature endothelium, a process known as sequestration. Sequestration causes micro vascular obstruction and compromises the blood flow through tissues such as the liver, spleen, lung, and brain. The effects of sequestration include mechanical obstruction (which can lead to hypoxia), metabolic disturbances and is a central point where parasite toxins and inflammatory mediators concentrate [47] (Figure 3).
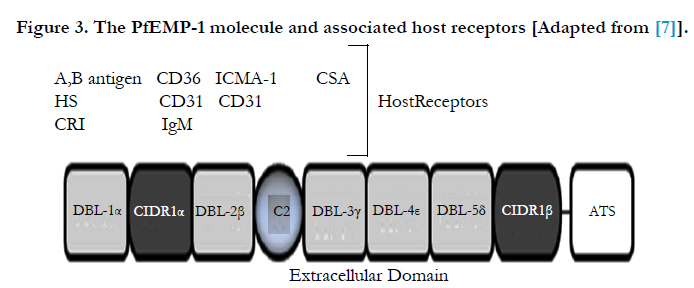
Figure 3. The PfEMP-1 molecule and associated host receptors [Adapted from [7]].
Increased expression of cytoadherence receptors enhances infected RBC sequestration to the endothelium via parasite derived proteins (expressed on the surface of the infected RBC), such as PfEMP-1. The principal parasite surface protein and sequestration ligand known as P. falciparum erythrocyte membrane protein 1 (PfEMP-1), encoded by var genes, is expressed. It is predominantly mature stage parasites (trophozoites and schizonts) that adhere to the microvasculature [16].
The PfEMP-1 molecule has a pivotal role in the pathogenesis of P. falciparum as a number of host receptors are recognized by the various extracellular binding domains of PfEMP-1. Thus permitting the infected RBCs to adhere with host endothelium. In the case of cerebral malaria, PfEMP-1 may mediate adhesion to several adhesion molecules, in particular ICAM-1 which is unregulated on the cerebral vascular endothelium [6].
The innate immune response is crucial to the outcome during a P .falciparum infection. Innate immune responses take effect immediately and provide an early defence until the adaptive immune response is engaged. In some cases, an infection by P. falciparum may be controlled by the innate immune system. 61 Parasite burdens observed in nonimmune individuals with acute P. falciparum malaria are lower than expected based on parasite replication rates observed in vitro, suggesting that the innate immune system can contribute to effective control of acute parasite replication before the adaptive immune response develops [34].
The innate immune system functions to limit the maximum parasite density, but gradually acquired adaptive mechanisms complete parasite elimination. The innate immune system is essential for most inflammatory responses that are triggered by monocytic cells, other leukocytes and mast cells through their innate sensing receptors. Macrophages are important in innate immunity as they are able to clear parasitized RBCs in the absence of opsonizing malaria-specific antibodies. It is hypothesized that there are two methods of infected RBC uptake by macrophages. The predominant method of uptake involves the binding of non-specific IgG and complement to the surface of infected RBCs, and increased exposure of senescent RBC markers such as exposure of phosphatidylserine (PS). This method induces the release of pro-inflammatory cytokines. The second method of uptake is CD36 mediated, which involves the binding of CD36 on the macrophage to PfEMP-1 on the infected RBCs. This method does not involve the release of pro-inflammatory cytokines [42, 20].
There are three main biochemical pathways that result in activation of the complement system: the classical complement binding pathway; the mannose-binding lectin pathway; and the alternative pathway. All three lead to the formation of C3 and C5 convertase which results in the cleavage of C3 and C5 into C3a, C3b, C5a and C5b, respectively. RBCs opsonized by IgG and complement (C3b) are recognized by the Fc receptor (FcR) and CR1 (respectively), and phagocytosed by macrophages. This method of clearance is effective in senescent and damaged RBCs, and also in P. falciparum infected RBCs [15].
CR1 is a 200-kDa single chain membrane bound glycoprotein and a member of the regulators of complement activation (RCA) gene cluster. CR1 possesses complex tri and tetra N-linked oligosaccharides in its mature form and the gene for this protein is located on the q32 arm of chromosome 1. It is composed of a number of repeated domains called short consensus repeats (SCRs) each of which is composed of 60 amino acids containing four invariant cysteines. The extracellular domain of the CR1 is composed of 30 SCRs, the first 28 of which are arranged in tandem repeats in homologous groups of 7, with each group known as long homologous repeat (LHR). SCRs 8-12 and SCRs 15-18 preferentially bind to C3b and SCRs 1-4 preferentially bind to C4b. The region of CR1 that interacts with infected erythrocytes to form rosettes has been mapped to LHRB and first three SCRs of LHR-C, SCR 10 and 17 have been particularly found to play an important role in this interaction [23, 22].
Differences in the expression of CR1 on erythrocytes might determine susceptibility of an individual towards development of cerebral malaria and severe malaria-associated anemia. In one of the studies it was suggested that young children may be more susceptible to SMA because of their lower levels of RBC complement regulatory proteins, which make them less equipped to handle IC formation and complement activation. Previously same group of researchers had proved that a decline in levels of CR1 and increase in immune complex levels significantly associates with SMA. The mechanism for the loss of CR1 from the surface of erythrocytes is being investigated. A series of experiments indicated that CR1 present in the form of clusters on RBC surface, undergoes unclustering due to the binding of IgM -C3b complexes to glycophorin A. Unclustering might promote rapid loss of CR1 from the surface of erythrocytes infected with the malaria parasite [7].
CR1 is a highly polymorphic glycoprotein. Three different polymorphic forms of CR1 have been identified, namely structural (size variation 160-250 kDa), density (high and low expression on RBCs controlled by alleles H and L) and knops blood group (McC (a+)/McC (b+); Sl (a+)/Sl (a-); Kna/ Knb).
Structural polymorphism: Four different structural polymorphic forms of CR1 are known, namely A, B, C and D (CR1*1, CR1*2, CR1*3, CR1*4) with respective molecular weights of 190, 220, 160 and 250 kDa (under non-reducing conditions). This polymorphism is regulated by four autosomal co-dominant alleles. A polymorphism in the CR1 transcripts with incremental differences of 1.4 kb in mRNA was present in donors expressing the various polymorphic forms. This difference corresponds to the size of one LHR and 40 kDa difference, seen among allotypic forms of CR1. Therefore on the basis of this observation it was suggested that the insertion or deletion forms the basis of structural polymorphism. Analysis of restriction fragment length polymorphism (RFLP) suggested that intragenic duplication rather than alternate mRNA splicing is responsible for the allotypic differences [24, 12].
Density polymorphism: Second type of polymorphism is a Hind III RFLP, which in Caucasians but not in Africans, correlates with CR1 copy number on erythrocytes. Homozygotes for the L (low expression) allele usually express fewer than 200 copies of CR1, homozygotes for the H (high expression) allele express several times this number and heterozygotes are intermediate. This polymorphism arises due to a single base change in the intron of d1d2 segment within the LHR-D (Long homologous repeat) region resulting in the generation of a polymorphic Hind III site within this region [8].
Genotypic frequencies of HH, HL and LL forms have also been studied in the malaria endemic and non-endemic groups in different populations. In nonendemic Caucasian and Choctaw population groups in USA, the gene frequencies for H and L alleles were found to be 0.82, 0.18 and 0.84, 0.16 respectively. In endemic Black Africans the gene frequencies for H and L alleles were 0.85 and 0.15; in S. Chinese-Taiwanese 0.71 and 0.29; in Pacific Asians 0.42 and 0.58 and in Cambodians 0.53 and 0.47 respectively [48].
Knops polymorphism: The third type of polymorphism represented by Knops blood group system is of particular interest. In this system, Mca and Mcb is one allelic antigen pair and Sla and Vil is another pair. The corresponding phenotypes for the first pair are McC (a+) and McC (b+) and for the second pair are Sl (a+) and Sl (a-). Studies have now established the molecular basis for Knops polymorphism. These antigens have been localized on the LHR-D segment of CR1. Single nucleotide polymorphisms occurring in SCR 25, which lead to amino acid substitutions, result in generation of these polymorphic forms Population based studies have been carried out to determine the distribution of different types of Knops polymorphic forms in different populations. The gene frequencies for Sl (a+) and Sl (a-) in African American persons are almost equal (0.48 vs. 0.52) wheras Sl (a-) is greatly increased in Africa [25, 33].
Out of the three polymorphic forms, size polymorphism has not been found to play a role in determining susceptibility to severe malaria. With regard to density polymorphism, some studies suggest that low-density allele confers protection against malaria, whereas another suggested that low- density allele might be a risk factor for severe forms of malaria. Erythrocytes with low CR1 expression (because of the homozygous LL genotype of CR1) have been shown to form reduced number of rosettes with Plasmodium falciparum infected cells [40, 36, 9].
The study of characteristics of immune responses against Plasmodium is based on murine experimental systems. Both, cell mediated and antibody-dependent immunity are required for adequate protection, likely encompassing different mechanisms finely tuned in time. Besides, innate immunity is thought to play a crucial role in clearing Plasmodium from parasitized hosts [34]. In splenic response, tissular changes that provoke alterations in blood flow through the organ. These changes prevent the access of infected erythrocytes to splenic tissues in which the immune response is going on until armed effector cells are produced. In general, most of the evidence supports the hypothesis that cells from the monocyte-macrophage lineage are more effective than neutrophils at phagocytosing parasitized erythrocytes [52].
Recent work has shown that P. chabaudi infection in γ/δ T-celldeficient mice have exacerbated early and chronic parasitemias. These results would fit well with data ascribing early production of gamma interferon (IFN-γ) and tumor necrosis factor alpha (TNF-α) both to spleenic ᵞ/ᵟ T lymphocytes and to natural killer (NK) cells [53, 41]. Both the cellular and humoral responses are pivotal elements in the eradication of Plasmodium from the body, and both are critically dependent on γ/δ CD4+ lymphocytes. It has been firmly established that CD4+ T cells are comprised of at least two functionally different subsets, distinguished on the basis of lymphokine secretion in Th1 (IFN-γ-producing) and Th2 (interleukin-4 [IL-4]/IL-5-producing) cells. CD4+ T cells of either Th1 or Th2 type also have regulatory functions in human P. falciparum malaria. Both Th1 and Th2 responses seem to be required to control the infection, but they need to be adequately tuned in intensity and time [26, 49].
The early production of IFN-γ to give resistance against infection. In support of this view, analysis of IFN-γ R-/- mice infected with P. chabaudi reveals a critical role of IFN-γ in immunity against this pathogen, although the production of parasite-specific immunoglobulins was not affected [Favre, N., et. al. 1997]. Interestingly, Tan et. al. reported that IFN responsive factor (IRF-1)-/- mice infected with P. berghei showed lower mortality than wild-type mice, although they produced no IFN-γ or NO [50].
It is likely that there are mechanisms of resistance independent of IFN- γ and NO. In this regard, it is interesting that treatment in vivo with anti-IFN-γ exacerbates P. yoelii 17XL infection in C57BL/6 because mice treated with antibody die earlier. In contrast, treatment with aminoguanidine, an irreversible inhibitor of NO production, has no effect. Consistently, mice lacking inducible nitric oxide synthase (iNOS-/-) cleared P. berghei XAT (an attenuated variant of P. berghei NK65) as effectively as did wildtype animals. In this case, resistance was dependent on IFN- γ, since it’s in vivo, blocking provoked progression of parasitemiaand death [53].
The overall conclusion that can be drawn from this is that the role of a particular cytokine is likely to be different at different stages of the infectious process. A prominent role in switching from Th1 to Th2 responses is attributed to IL-10. Therefore, it is probably involved in controlling the adequate timing of antiparasitic responses. Early IL-10 production has been associated with susceptibility to infection, and it is thought that this cytokine has a prominent anti-inflammatory effect, limiting in some way the damage inflicted on normal tissues by an excessive Th1 response [27, 54].
The Th2 profiles have been reported in humans, with elevated levels of IgE being found in the blood of malaria patients, presumably due to the predominance of Th2 cells over Th1 helper cells. This polarization was significantly higher in the case of patients suffering from severe malaria [39]. Th1 responses are important for clearance of P. falciparum malaria. Non-immune children with severe P. falciparum malaria showed lower levels of IL-12 and IFN-γ in serum and had a reduced capacity to produce them after in vitro stimulation. It is interesting that children with severe anemia had the highest levels of TNF-γ [28]. Furthermore, Luty et al. [29] found that peripheral blood mononuclear cells of patients with mild malaria produced IFN-γ in response to malarial antigens, whereas those with severe malaria did not. The studies on Ghanaian childrenshowed that only patients with uncomplicated malaria had a positive correlation with levels of TNF- α and soluble TNF- α R1 and TNF- α R2 in serum. In the same study, children with CM had high levels of TNF- α, and although TNF- α level were associated with fever no differences were observed in soluble TNF- α receptors. Interestingly, children with fever and detectable parasitemia, but not afebrile parasitized patients, had elevated levels of TNF- α [35]. In this regard, patients who died from P. falciparum malaria had higher amounts of IL-6, IL-10, and TNF- α in serum than did the patients who survived. In the present study, IL-6, IL- 10, and IFN- γ were associated with hyperparasitemia, jaundice, and shock. Moreover, TNF- α was associated with renal failure. Surprisingly, lower levels of cytokines were found in CM. In an important finding, a relative deficiency of IL-10 was detected as death approached [10].
Conclusion
Malaria is a worldwide spreaded disease due to plasmodium species (P. falciparum, P. vivax, P. malariae and P. ovale). It affects 300- 500 million people in which 1-3 million going to death. All of plasmodium species, P. falciparum is very dangerous leads to cerebral malaria. For completing, his life cycle, plasmodium having two host first mosquito (as a vector) and second human. When plasmodium is introduced in blood by mosquito, it attached with RBCs through CD35 (CR1) which act as a receptor for PfEMP-1 (present in plasmodium) and circulate in blood stream. CD35 is a cell surface receptor and its gene present in chromosome no. 1. It is also known as Complement Receptor Type 1 (CR1). Density of CD35, in cell surface is determined by CR1 gene polymorphism. There are types of CR1 polymorphism (a) Structural polymorphism (b) density polymorphism (c) knops polymorphism. High density of CD35 is more susceptible to plasmodium infection. ICAM-1 and VCAM-1 are also play important role in malarial pathogenesis. Cytokine profiling indicate malarial severity. Pro-inflammatory cytokine TNF-α and INF- γ level is increased during malarial infection where as IL10, IL4, and IL6 are anti-inflammatory cytokine.
References
- Ahearn JM, Fearon DT (1989). "Structure and function of the complement receptors, CR1 (CD35) and CR2 (CD21)". Adv. Immunol. 46: 183–219.
- Baum J, Maier AG, Good RT, Simpson KM, Cowman AF (2005). Invasion by P. falciparum merozoites suggests a hierarchy of molecular interactions. PLoS Pathog. 1:e37.
- Cavasini, M. T. Ribeiro, W. L. Kawamoto, F. and Ferreira, M. U (2000). “How prevalent is Plasmodium malariae in Rondonia, western Brazilian Amazon?” Revista da Sociedade Brasileira de Medicina Tropical, vol. 33, no. 5, pp. 489–492.
- Carter R, Mendis KN (2002). Evolutionary and historical aspects of the burden of malaria. Clin Microbiol Rev. 15:564-594.
- Cowman AF, Crabb BS (2006). Invasion of red blood cells by malaria parasites. Cell.;124:755-766.
- Cserti CM, Dzik WH (2007). The ABO blood group system and Plasmodium falciparum malaria. Blood. 110:2250-2258.
- Craig ML,Waitumbi JN, Taylor RP (2005). Processing of C3bopsonized immune complexes bound to non-complement receptor 1 (CR1) sites on red cells: Phagocytosis, transfer and associations with CR1. J Immunol 174:3059-66.
- Cornillet P, Philbert F, Kazatchkine MD, Cohen JH (1991). Genomic determination of the CR1 (CD35) density polymorphism on erythrocytes using polymerase chain reaction amplifi cation and HindIII restriction enzyme digestion. J Exp Med; 136:193-7.4
- Cockburn IA, Mackinnon MJ, O’Donnell A, Allen SJ, Moulds JM, Rowe JA, et al. (2004) A human complement receptor 1 polymorphism that reduces Plasmodium falciparum rosetting confers protection against severe malaria. PNAS; 101:272-7.
- Day, N. P., Hien T. T., Schollaardt T, Loc P. P., Chuong L. V., et al (1999). The prognostic and pathophysiologic role of pro- and antiinflammatory cytokines in severe malaria. J. Infect. Dis. 180:1288–1297.
- Desimone TM, Jennings CV, Bei AK, et al (2009). Cooperativity between Plasmodium falciparum adhesive proteins for invasion into erythrocytes. Mol Microbiol.
- Dykman TR, Hatch JA, Aqua MS, Atkinson JP (1985). Polymorphism of the C3b/C4b receptor (CR1): Characterization of a fourth allele. J Immunol; 134:1787-9.
- Favre, N., Ryffel B., Bordmann G., and Rudin W. (1997). The course of Plasmodium chabaudi chabaudi infections in interferon-γ receptor deficient mice. Parasite Immunol. 19:375–383.
- Greenwood BM, Fidock DA, Kyle DE, et al (2008). Malaria: progress, perils, and prospects for eradication. J Clin Invest. 118:1266-1276.
- Guo RF, Ward PA (2005). Role of C5a in inflammatory responses. Annu Rev Immunol.;23:821-852.
- Horrocks P, Pinches RA, Chakravorty SJ, et al (2005). PfEMP1 expression is reduced on the surface of knobless Plasmodium falciparum infected erythrocytes. J Cell Sci. 118:2507-2518
- Hay SI, Guerra CA, Tatem AJ, Noor AM, Snow RW (2004). The global distribution and population at risk of malaria: past, present, and future. Lancet Infect Dis. 4:327-336.
- Hume JC, Lyons EJ, Day KP (2003). Human migration, mosquitoes and the evolution of Plasmodium falciparum. Trends Parasitol. 19:144-149.
- Idro R, Jenkins NE, Newton CR (2005). Pathogenesis, clinical features, and neurological outcome of cerebral malaria. Lancet Neurol.; 4:827-840.
- Janeway CA, Jr., Medzhitov R (2002). Innate immune recognition. Annu Rev Immunol.;20:197-21 Hume JC 6.
- Kwiatkowski D, Hill AV, Sambou I, et al (1990). TNF concentration in fatal cerebral, non-fatal cerebral and uncomplicated Plasmodium falciparum malaria. Lancet. 336:1201-1204.
- Klickstein LB, Bartow TJ, Miletic V, Rabson LD, Smith JA, Fearon DT (1988);. Identifi cation of distinct C3b and C4b recognition sites in the human C3b/C4b receptor (CR1, CD35) by deletion mutagenesis. J Exp Med 1988; 168:1699-717.
- Krych M, Hourcade D, Atkinson JP (1991). Sites within the complement C3b/C4b receptor important for the specifi city of ligand binding. PNAS 88:4353-7.
- Katyal M, Sivasankar B, Ayub S, Das N (2003). Genetic and structural polymorphism of complement receptor 1 in normal Indian subjects. Immunol Lett 89:93-8.
- Krych-Goldberg M, Moulds JM, Atkinson JP (2002). Human complement receptor type 1 (CR1) binds to a major malarial adhesin. Trends Mol Med 8:531-7.
- Kobayashi, F., T. Morii, T. Matsui, T. Fujino, Y. Watanabe, et al (1996). Production of interleukin 10 during malaria caused by lethal and nonlethal variants of Plasmodium yoelii yoelii. Parasitol. Res. 82:385–391.
- Linke, A., R. Kuhn, W. Muller, N. Honarvar, C. Li, and J. Langhorne. (1996). Plasmodium chabaudi chabaudi: differential susceptibility of genetargeted mice deficient in IL-10 to an erythrocytic-stage infection. Exp.Parasitol. 84:253–263.
- Luty, A. J., Perkins D. J., Lell B., Schmidt-Ott , R., Lehman L. G., et al (2000). Low interleukin-12 activity in severe Plasmodium falciparum malaria. Infect. Immun. 68:3909–3915.
- Luty, A. J., B. Lell, R. Schmidt-Ott, L. G. Lehman, D. Luckner, et al (1999). Interferon- γ responses are associated with resistance to reinfection with Plasmodium falciparum in young African children. J. Infect. Dis.179:980–988.
- Moulds JM, Nickells MW, Moulds JJ, Brown MC, Atkinson JP (May 1991). "The C3b/C4b receptor is recognized by the Knops, McCoy, Swainlangley, and York blood group antisera". J. Exp. Med. 173 (5): 1159–63.
- . Miller LH, Baruch DI, Marsh K, Doumbo OK (2002). The pathogenic basis of malaria. Nature. 415:673-679.
- Mayer DC, Kaneko O, Hudson-Taylor DE, Reid ME, Miller LH(2001). Characterization of a Plasmodium falciparum erythrocyte-binding protein paralogous to EBA-175. Proc Natl Acad Sci U S A. 98:5222-5227.
- Moulds JM, Thomas BJ, Doumbo O, Diallo DA, Lyke KE, Plowe CV, et al (2004). Identifi cation of the Kna/Knb polymorphism and a method for Knops genotyping. Transfusion;44:164-9.
- Mohan, K., and M. M. Stevenson, (1998). Acquired immunity to asexual blood stages in malaria, p. 467–493. In I. W. Sherman (ed.), Parasite biology, pathogenesis, and protection. ASM Press, Washington, D.C.
- McGuire, W., D’Alessandro U., Stephens S., Olaleye B. O., Langerock P., et al (1998). Levels of tumour necrosis factor and soluble TNF receptors during malaria fever episodes in the community; Trans R Soc Trop Med Hyg.Jan-Feb;92(1):50-3.
- Nagayasu E, Ito M, Akaki M, Nakano Y, Kimura M, Looareesuwan S, et al (2001). CR1 density polymorphism on erythrocytes of falciparum malaria patients in Thailand. Am J Trop Med Hyg 64:1-5.
- Newton CR, Hien TT, White N. Cerebral malaria (2000). J Neurol Neurosurg Psychiatry.; 69:433-441.
- Naik, R.S., Branch O.H., Woods A.S., Vijaykumar M., Perkins D.J (2000). Glycosylphosphatidylinositol anchors of Plasmodium falciparum: molecular characterization and naturally elicited antibody response that may provide immunity to malaria pathogenesis. J. Exp. Med. 192:1563–1576.
- Perlmann, P., Perlmann H., ElGhazali G., and Blomberg M. T. (1999). IgE and tumor necrosis factor in malaria infection. Immunol. Lett. 65:29–33.
- Rowe JA, Rogerson SJ, Raza A, Moulds JM, Kazatchkine MD, Marsh K, et al (2000). Mapping of the region of complement receptor 1 (CR1) required for Plasmodium falciparum rosetting and demonstration of the importance of CR1 in rosetting in fi eld isolates. J Immunol 165:6341-46.
- Seixas, E. M., and J. Langhorne. (1999). ᵞ/ᵟ T cells contribute to control of chronic parasitemia in Plasmodium chabaudi infections in mice. J. Immunol. 162:2837–2841.
- Serghides L, Kain KC (2001). Peroxisome proliferator-activated receptor gamma-retinoid X receptor agonists increase CD36-dependent phagocytosis of Plasmodium falciparum-parasitized erythrocytes and decrease malaria-induced TNF-alpha secretion by monocytes/macrophages. J Immunol.166:6742-6748.
- Sim BK, Chitnis CE, Wasniowska K, Hadley TJ, Miller LH (1994). Receptor and ligand domains for invasion of erythrocytes by Plasmodium falciparum.Science. 264:1941-1944.
- Snow RW, Guerra CA, Noor AM, Myint HY, Hay SI (2005). The global distribution of clinical episodes of Plasmodium falciparum malaria. Nature.434:214-217.
- Tuteja R. (2007) Malaria - an overview. Febs J. 274:4670-4679.
- Trampuz A, Jereb M, Muzlovic I, Prabhu RM (2003). Clinical review: Severe malaria. Crit Care. 7:315-323.
- Tumer AR, New bold GD, (1994) Cerebral malaria: the sequestration hypothesis. Parasitol Today.; 10:412-414.
- Thomas BN, Donvito B, Cockburn I, Fandeur T, Rowe JA, Cohen JH, et al (2005). A complement receptor 1 polymorphism with high frequency in malaria endemic regions of Asia but not Africa. Genes Immun; 6:31-6.
- Torre, D., Speranza F., Giola M., Matteelli A., Tambini R., and Biondi G. (2002). Role of Th1 and Th2 cytokines in immune response to uncomplicated Plasmodium falciparum malaria. Clin. Diagn. Lab. Immunol.9:348–351.
- Tan, R. S., Feng C., Asano Y., and Kara A. U. (1999). Altered immune response of interferon regulatory factor 1-deficient mice against Plasmodium berghei blood-stage malaria infection. Infect. Immun. 67:2277–2283.
- WHO (2000), Communicable Diseases Cluster. Severe falciparum malaria. Trans R Soc Trop Med Hyg. 94 Suppl 1:S1-90.
- Yadava A., Kumar S., Dvorak J. A., Milon G., and Miller L. H (1996). Trafficking of Plasmodium chabaudi adami-infected erythrocytes within the mouse spleen. Proc. Natl. Acad. Sci. USA 93:4595–4599.
- Yoneto, T., Yoshimoto T., Wang C. R., Takahama Y., Tsuji M., Waki S., and Nariuchi H. (1999). Gamma-interferon production is critical for protective immunity to infection with blood-stage Plasmodium berghei XAT but neither NO production nor NK cell activation is critical. Infect.Immun. 67: 2349–2356.
- Yoshida A., Maruyama H., Kumagai T., Amano T., Kobayashi F, et al (2000). Schistosoma mansoni infection cancels the susceptibility to Plasmodium chabaudi through induction of type 1 immune responses in A/J mice.Int. Immunol. 12:1117–1125.





