The Use of Medical Devices in Italy And Europe: The Need Of A Careful Control Policy
Vacchiano G1, Vyshka G2*
1 Associated Professor of Legal Medicine; University of Sannio, Benevento, Italy.
2 Associated Professor, Biomedical and Experimental Department, Faculty of Medicine, University of Tirana, Tirana, Albania.
*Corresponding Author
Gentian Vyshka,
Associated Professor,
Biomedical and Experimental Department,
Faculty of Medicine, University of Tirana,
Rr Dibres 371, Tirana, Albania.
Tel: +355692828140
Fax: +3552362710
E-mail: gvyshka@yahoo.com
Article Type: Review Article
Recieved: February 19, 2015; Accepted: March 26, 2015; Published: March 26, 2015;
Citation: Vacchiano G, Vyshka G (2015) The Use of Medical Devices In Italy and Europe: The Need of a Careful Control Policy. Int J MedBiotechnol Genetics. 03(2), 21-26. doi: dx.doi.org/10.19070/2379-1020-150004.
Copyright: Vyshka G© 2015. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
Abstract
The use of medical devices is in continuous expansion and constitutes a valid aid to ailing people, due to remarkable technological advances. The regulation on their circulation in Italy and Europe are inspired by the principle of free circulation. In fact it is sufficient for a device to have the CE logo for it to be freely commercialized in all of Europe.
This way of operating that favours commerce, exposes the sick to the risk of harm from defective devices that have not been suitably checked prior to their commercialization. Having discussed a few episodes characterized by device malfunctioning, the authors observe the need for a more careful “control” policy for devices, adequate care on the part of doctors who use such tools, and of suitable insurance measures for the patient in the event of harm.
2.Definition of The Issue
3.Discussion
4.References
Introduction
The growing and unrelenting need to improve quality of life, and the will to ensure the best possible health conditions for Human Beings, have over the years nurtured the ever increasing interest for medical devices, broadly defined as devices designed to be used on Humans to improve their health and quality of life. Furthermore, the remarkable scientific and technological advances, applied to biology, have permitted an ever increasing number of sophisticated and inventive solutions to properly counter and overcome disablements, handicaps and pathologies that were once considered to be incurable, and there is an increasing demand for medical devices from a large number of people. As defined by a specific European Union [1] directive, a medical device is defined as any instrument, device, installation, software, substance or othother product, used alone or in combination, including accessories, that is designed by the manufacturer to be used on humans for:
• Diagnosis, prevention, control, treatment or alleviation of disease.
• Diagnosis, control, treatment, alleviation or compensation for a lesion or handicap.
• Study, substitution or modification of the anatomy or of a physiological process.
• Birth control
Definition of The Issue
The list of devices does not include those tools whose effect in the body or on the body takes place via pharmacological or immunological mechanisms or by metabolic processes whose function can be enhanced by such tools. This is an extremely wide definition that includes not only prostheses and devices designed to be implanted and remain in the body more or less permanently, but also reagents, equipment and accessories used to detect clinical, morphological and biohumoral parameters, as well as substances used for disinfecting wounds, aides for the disabled and healthcare devices in general. The number of medical devices is therefore very large, in continuous expansion and their use, as a result of substantial social necessities, involves a vast proportion of the population. While at the beginning of the century [2], only four classes of devices were recognized in Italy (pessaries-irrigators, showers, syringes, cannullae-disinfectants – devices for containing hernias), today medical devices are grouped, following the directive of a Specific Commission [3], by usage modality, and according to the type of anatomical-functional modifications induced. Moreover they are further divided into 21 categories (Table. 1), and in four ascending risk classes, which are based on their interaction with biological structures or vital functions (Table. 2).
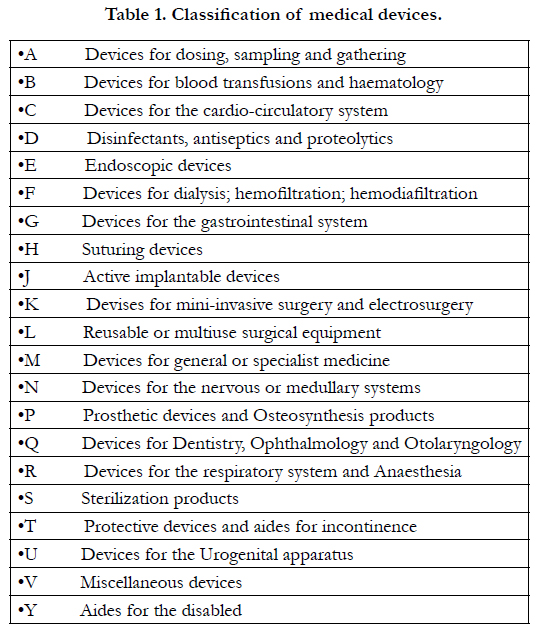

Table 1. Classification of medical devices.
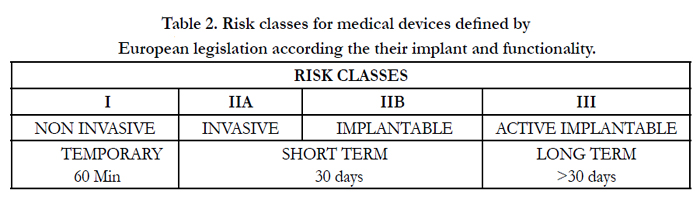
Table 2. Risk classes for medical devices defined by European legislation according the their implant and functionality.
The availability of a vast number of devices, their potential hazards together with their increasing use by a large number of individuals, induced the European [4] and Italian [5] legislators to pass a considerable number of laws aimed at insuring mandatory safety standards and effectiveness with regards to design, building, choice of material and absence of harmful substances, while assuring the free circulation on the European market of devices that respect these characteristics [6]. The corresponding certification, confirming the attainment of the specified criteria through a specific symbol (CE), is emitted by an Independent Organization monitored by the National Authority [7]. Therefore a new procedure has become established for the necessary checks and authorizations needed for the commercialisation of any given device. While under the traditional system, there was a penetrating form of control on the part of the National Authority, today with the onset of the European Community a more liberal approach has become prevalent based on the freedom – responsibility doctrine [8]. Individual device manufacturers must respect the predefined safety and efficacy standards through an adequate clinical trial process. National Authorities are responsible for monitoring the devices on the market and Independent Organizations for granting conformity certification, however they cannot prevent the free circulation of the devices within the Community, unless in particular cases to request further steps, checks and verifications [9].
Such a solution, while doubtlessly much appreciated by the industrial world because the commercialization times for devices are noticeably shortened, shows all its vulnerability in entrusting the ethical, political and social sensibility of the manufacturers and Independent Organizations with compliance to the fundamental safety prerequisites provided for by the European Community [10]. Specific European and Italian legislation, however, make it mandatory to report accidents or missed accidents in the use of medical devices, for appropriate monitoring in the framework of a surveillance system aimed at increasing the level of protection for patients and medical operators and avoid similar accidents reoccurring after any length of time (Table. 3).

Table 3. Procedure to be followed in case of device accidents or missed accidents.
The monitoring system that is structured through the assessment of the accidents reported and their appropriate disclosure, allows for the National Authority not only to request modifications to the device if it so wishes, but also order it to be withdrawn from the market if it is considered to be hazardous to safety and health [12]. In fact the use of implantable medical devices, whether active or not, and of certain electro-medical equipment, is not devoid of difficulties and hazards, either because the device is very sophisticated, or because of the pathological substrate, or because of delicate and sometimes laborious implantation procedure, and this can sometimes force the operator to make difficult decisions regarding the cost benefit evaluation. Preliminary consideration however must be given to the fact that the ideal device, in other words a device that optimally replaces altered anatomical structures without any local or general organism level interaction, and considerably improves the compromised functionality, is still not available. Furthermore, the availability of a considerable number of devices for the same organ or anatomical structure frequently places medical operators in front of though choices because they must be very careful to chose the most suitable and effective device.
So the selection of the device to implant is directed either by some distinctive characteristic of the device that are thought to be particularly useful for the specific pathological situation or by the patient’s pathological condition that requires the use of a specific device, excluding others.
In some individuals it is possible to implant mechanical valvular cardiac devices, while biological valves that do not require anticoagulants, but are characterized by a limited functional reliability and progressive failure [13], are more suitable for other patients.
Analogously it is possible to operate on the coxo-femoral bone structures by implanting a wide variety of prosthesis of different forms and materials (steel, chrome-cobalt, titanium) that may or may not require the use of PMMA and that can also be built combining different materials (metal/polyethylene; ceramic/polyethylene).
Of course, the quality of the bone structures, the acetabular and femoral morphology and the age of the patient will condition the choice of the prosthetic system and fixing method to be adopted [14]. The device to be employed must, however, be in compliance with the directives of the specialized Literature, as the use of dated devices, or ones with unsatisfactory guarantees of success cannot be excused. In synthesis choosing the device to implant or to use is a moment of special attention because the medical operator must undertake a punctual and precise assessment able to ponder a whole array of parameters, some of which can be determined from the patient, others relating to the device, which is of crucial importance for the success of the implant. The choice of the device must therefore be particularly careful in assessing the risk/benefit relationship that must be advantageous to the patient, which should be informed and involved in the choice, not so much for the necessary consent procedure, but also and above all to get to know the characteristics of the device and the necessary checks that need to be carried out. Of course the medical operator can make the choice of device in full freedom based upon his or her knowledge, technical skills, experience and specialist training, but also baring well in mind the scientific knowledge and data from the literature, given that the device must necessarily meet those assessment parameters described and reported above.(Table. 4)
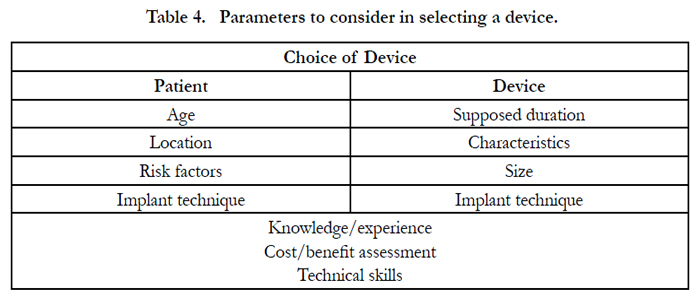
Table 4. Parameters to consider in selecting a device.
Implanting the device requires further care because of the possible negative consequences that may derive if the correct approach is not followed and if adequate expertise, experience and professionalism are not exercised. Having installed such devices, it is of mandatory importance to exercise particular care in monitoring and checking on the clinical conditions of the patient for a congruous time frame, especially if the application of the device proves difficult and if it involves particularly important structures that may compromise the life of the patient if damaged, as occurs for example in the case of lesions to major blood vessels [15]. Recent recommendations issued by the Italian Government [16] stress the necessity of a suitable, adequate maintenance of medical devices and of electro-medical equipment, which would constitute an improvement to an obsolete concept, aimed only at repairing malfunctioning devices, and would concentrate instead at preventing risk and guaranteeing service quality. The suggestion is for the institution of a Service, within every Health Authority, designated to managing and maintaining the biomedical technological assets, as well as developing and implementing adequate technical ability.
Such recommendations, which are fully acceptable, receive further support from a specific recent ruling of the Italian Supreme Court [17] that passed judgement on the case of a newborn baby who died as a result of a malfunction of an incubator whose sensors were not calibrated, causing the internal environment to overheat. The Court of Cassation on that occasion, in line with previous judgements from the Benevento Tribunal [18] and the Naples Court of Appeals [19], not only condemned the Manager of the Operative Unit for having tolerated...the use of an incubator that was not included in the programmed maintenance scheme...because he....should have been vigilant to constantly ensure the perfect functioning of the incubator, but also the managing directors of the Hospital Authority because...they were promoters of an irregular practice with regards to the maintenance of electro-medical equipment in as there was complete discretional power on the part of the Technical Office with regards to subjecting machinery to programmed or requested maintenance. So in some cases electromedical machinery, though outdated, were sheltered from any maintenance for periods in excess of a year. In synthesis, a much needed and pressing control and maintenance activity is necessary for medical devices and electro-medical equipment so that their use can take place safely, granting the patient and the medical operator the necessary guarantee of relaxed operating conditions. And such an activity would fully and suitably involve not only the individual operators and the Maintenance Service Staff, but also the Authority Management in monitoring this task and insuring the standards that guarantee that this action be carried out in the best possible way. However, even when the medical operator behaves in a sensible way in the choice and installation of the device and its correct and adequate use by the patient, who undergoes the necessary checks, the suggested therapies and the most appropriate hygiene practices, the application of a device doesn’t always offer the benefits required and expected because of malfunctioning. This is a possibility that, however, scares and arouses much disappointment because of the serious risk for health and the life of the patient deriving from this. The Specialized Literature has advised on the existence of defective cardiac valves [20] and recently a cardiac valve was reported in Italy, in which the pivot of the metal disk would frequently brake, putting the patient’s life in serious danger. Out of 36 valves implanted 5 deaths were reported and 12 patients had to have the defective valve removed and replaced [21].
Subsequently, the less than perfect “duration” of aortal endoprostheses [22] due to the use of unsuitable materials was reported as well as the clouding of artificial crystalline lenses caused by the wear of the polymers and the presence of residual silicone on their surface [23] to the extent that following the recommendation of the Soc. Oftalmologica Italiana the substitution of the implanted devices was prudently adopted. Furthermore, with regards to hip prosthesis, alterations in the high molecular weight polyethylene that because of an early oxidisation caused by X-rays during sterilisation would brake [25], causing the failure of the implant. The Ministry of Health, commenting on this, recommended not to use X-ray sterilized prostheses or ones that were poorly stored [26]. Some mammary prostheses based on soy oil have also been found to be potentially dangerous for health. In fact, because of capsular degradation, body lipids could come into contact with soy oil generating genotoxic aldehydes. These prostheses were withdrawn from the market and subsequent directives from the M.D.A. suggested their removal since phenomena of hyperplasia, metaplasia, and presence of refrigerating materials within the capsule, as well as a flourishing reaction to the foreign body were reported. Finally, (Table. 5) it is worth reporting the possible ruptures that can occur on mammary prostheses that are correlated to the age of the implant and unrelated to the technique used [28]. A Swedish study that examined 106 women, who had been implanted with 109 prostheses reports that after 11 years the percentage of breakage was about 8%.

Table 5. Breast implants, implant’s age and breakage.
Analogously with such results, a Danish study monitored and analysed 533 prosthetic mammary implants, between 1973 and 1997, reports that after 16-20 years from implantation, the percentage of malfunctioning prostheses is around 26% [30].
Recently, on the basis of an opinion expressed by the Superior Health Council, some mammary prostheses have been withdrawn from the market in Italy (Poly Implant Prostheses) because the material was not in conformity with the required standards. A census of these mammary prostheses has been started. The patients that carry these prostheses have been invited to discuss their situation with their surgeon and the cost of any removal has been taken on board by the SSN. Furthermore, with dedicated law a national register of mammary prosthetic implants has been instituted and mammary plastic surgery has been banned for underage women. [31]. These examples reported in the literature that relateto the sub-optimal functionality of medical devices implanted and the measures adopted, necessarily lead to a consideration on the need of suitable, proper checks in the planning, building and experimental phase of the device.
Discussion
The current system needs suitable corrective measures, given that merely placing a CE stamp cannot represent the only permit for the commercialization of every device. The directives of the European Community rightly highlight the need for a careful invigilation to avoid the CE stamp being used illegally and point out the possibility of imposing further discretional National regulations, so as to best guarantee the health and safety of patients and operators [32]. In addition, it seems necessary to increase necessary checks on the various Notified Bodies by the National Authority, which is called upon to make sure the system works correctly [33], and the institution of appropriate measures, such as national registers for devices, to monitor implanted devices over time as documented for example by validated experiences relating to hip prostheses [34]. Such a measure should in our opinion be adopted for all devices wether active or not. Finally, to protect patients from being damaged by defective medical devices, a careful premarket evaluation of medical devices and a severe identification of manifacturers, like in US happen [35], should be ensured; besides an insurance policy should be provided and paid for by the manufacturing firms, or rather, a guarantee fund should be established to finance any damage caused by defective medical devices, within the European Community or within individual states. After all, the conception, manufacturing and commercialization of medical devices, above all of implantable ones can be identified as a dangerous activity, given the inherent and not negligible danger to life and health that their malfunctioning may cause. So, similarly to what has been ratified by Italian Jurisprudence with regards to the production of hemo-derivatives [36], in the planning and manufacturing of a device, the manufacturer is called upon not only to abide by the European Norms, but also to offer guarantees of having worked with the “maximum prudence and diligence”. After all, in the event of a malfunction of a medical device the presumable responsibility of the manufacturer is often not recognised as a result of the difficulty in proving that the product was defective [37].
The regulation on this provided for in the Italian Code (art. 120 Cod. Cons.; art. 2697 CC) envisage the duty on the part of the damaged party to prove the existence of a defect in the device, the damage sustained as a result and the causal nexus between defect and damage. These often represent an insurmountable challenge for the patient because of the extreme difficulty of proving the defect in the device, given that the “defect” can be related to the conception, manufacturing, functionality and use of the device. Proof, of itself very hard to find, especially when the medical device is implanted, often becomes practically impossible. In fact a medical device that is removed following malfunction is hardly ever examined, is not available to the patient, and can be destroyed during the removal, so the proof of any “defect” is lost. So much so that authoritative doctrine has suggested that the burden of proof, ratified by the Consumer Code (art. 118) should be inverted, forcing the manufacturer to prove all the circumstances needed to exclude liability [38].
The Italian Court of Cassation [39] was called upon to rule on the “emptying” of a mammary prosthesis that occurred shortly after it was implanted and despite the absence of a specific proof of any “defect” in the device, it ruled that the emptying of the prosthesis was the result of a possible defect in the device. The court established that “the use of the product determined abnormal results compared to the normal expectations and such as to highlight the existence of a defect”. If however, in this case, because it regarded an implantable device, such a ruling can be basically approved, it is equally true that in the case of medical devices that require ample collaborative autonomy on the part of the patient for their correct functioning (i.e. a dental prosthesis), the proof that the desired result was not achieved and of the device not working optimally is not in of itself the proof of the defectiveness of the device. On the topic of malfunctioning medical devices, there has also been a recent contribution from the European Court of Justice, called on to rule on a dispute between the Heraklion Hospital and Medipac, a firm that had won a public competition to supply surgical suturing materials [40]. Because the hospital’s surgeons noticed that the materials and devices provided by the firm, though in possession of the CE certification, produced unsatisfactory and unreliable results, the offer was rejected and the contract was not stipulated. A controversy followed that reached the attention of the European Court of Justice. The court established that the free circulation of devices should be reconciled with the protection of patient’s health and that safeguarding Public Health constitutes a predominant requirement of public interest that confers the right for member states to disregard the free movement of goods as long as the measures adopted observe the principle of proportionality. Consequently the right for the Hospital to adopt all measures necessary for the procurement of necessary medical devices was recognised. This ruling takes on a distinctive relevance in as much as it stresses the necessary safeguarding of public health and the necessary and much needed safety and effectiveness guarantees to be offered to patients and medical operators, as it is not possible for economic considerations to become the main if not the only parameter in the choice of a device.
References
- Guideline 2007/47/CE del 5 Set. (2007). That modifies the 90/385/CE guideline on the convergence of the Member State Legislation relating to active implantable medical devices, the 93/42/CEE guideline on medical devices and the 98/8/CE guideline on the commercialization of biocides
- Refer to the L. 23 Giugno 1927 n. 1070 later converted in the R.D. 27 Luglio 1934, n. 1265 (Single Reference text of Medical Law).
- The Unified Commission for Medical Devices established by L.27 Dic. 2002 art. 57, provides for the classification of all medical devices on the market in Italy defined by art. 1 of D.L. n. 507, 14 December 1992 relating to guideline 90/385/CEE and art. 1 of D.L. n. 46, 24 December. (1997) relating to guideline 93/42/CEE, the national classification of medical devices currently in force was emanated by Decree of the Ministry of Health on 20 February 2007
- European guidelines on medical devices Dir 90/385/CEE, Dir. 93/42/CEE, Dir. 2003/12/CE, Dir. 2003/32/CE, Dir. 2003/50/CE, Dir. 2007/47/CE
- D.M. Sanità 23 July 1998, D. M. Sanità 10 October 2001, D.M. Salute 3 February 2003, D. M. Salute 5 August 2005, D. M. Salute 15 November 2005, L.23 December 2005 n. 266, L. 27 December 2006 n. 296, D.M. Salute 20 February 2007
- Ministry of Health, General Management of Pharmaceuticals and Medical Devices: Dispositivi medici (aspetti regolatori e operativi) Roma 2007
- For the list of accredited Authorities in Europe refer to: ec.europa.eu/enterprise/sectors/rtte/files/en98-13_en.pdf
- M. Barni Medical devices : the rules , the doubts , the risks. (2005) Profession, 2 , 6-11
- Requirements, classification and procedures for Conformity accreditation are in: Dispositivi medici (aspetti regolatori e operativi), Direzione Generale Farmaci e Dispositivi medici- Ministero della Salute - Roma 2007
- Ibid, 8.
- Refer to: Dir. 90/385/CEE from the 20 June 1990 art. 8, Dir. 93/42/CEE from the 14 June 1993 art. 10; D.L. 14 December 1992 n. 507 art.11; D.L. 24 February 1997 n. 46 art. 9 and 10; MEDDEV 2,12-1 Rev.4 . April 2001; Nota Min. 27 July 2004; Nota Min. 28 July 2004; D. Min della Salute 15 November 2005.
- Monitoring and vigilance on medical devices in : Medical ( regulatory aspects and operational) - Directorate General of Drugs and Medical Devices - Ministry of Health - Rome 2007
- Cotufro M, Renzulli A, Vosa C, Cerasuolo F, De Santo L. Vacchiano G, et al (2003). Protesi valvolari cardiache in Le protesi dalla Clinica alla Medicina Legale (a cura di Luigi Palmieri) Minerva Medica Ed. Torino.
- Guida G, Ronca D, Gatto S, Vacchiano G, Fedeli P, et al. (2003) Apparato locomotore in Le protesi dalla Clinica alla Medicina Legale (a cura di Luigi Palmieri) Minerva Medica Ed. Torino.
- Harris PL, Vallabhaneni SR, Desgranges P (2000 ) Incidence and risk factors of late rupture, conversion, and death after endovascular repair of infrarenal aortic aneurysms: the Eurostar experience. J.Vasc Surg 32 :739-49
- Minister of Social Affairs ((Sep 2008)) Recommendation for the prevention of adverse events resulting from the malfunction of the medical devices /medical equipment . Recommendation no . 9 .
- Supreme Court IV Sec . Pen. Sent. No. 1753 of December 5, 2007
- Trib . Benevento Sent. No. 1240 of March 10, 2000
- Court of Appeal of Naples Sent. No. 3464 of April 30, 2004
- Hansen L, Danne M, Hoffmann B, Riess F.C (2007) Structural valve failure with every beat regurgitation observed using the Medtronic Advantage aortic valve – J. Thorac Cardiovasc. Surg 134: 1344-1345
- Bottio T, Casarotto D, Thiene G, Caprili L, Angelici A, et al. (2003) Leaflet escape in a new bileaflet mechanical valve TRI technologies - Circulation 13 1-4
- Veith FJ, Baum RA, Ohki T (2002) Nature and significance of endoleaks and endotension: summary of opinions expressed at an international conference.J. Vasc. Surg 35:1029-35.
- Mattova J, Bahacava F, Murgasova Z (2004) Opacification of hydrophilic Memorylens U940A intraocular: analysis of 2 explanted lenses –J Cataract Refract Surg 30:193-9
- Soc . Oftamologica Italian : Press to all ophthalmologists Italian . Subject: Calcification - discoloration crystalline . Prot . N . 01630 / S / 03 of September 18, 2003
- Torre M (2005) Polietilene ad elevato peso molecolare per protesi ortopediche. Sterilizzazione, degrado e usura. Not. Ist. Super. Sanità 18, 3-8
- Min . Of Health , Dep . Innovation , Gen. Dir. Of Medicines and Medical Devices Bureau III (Mar 2005). Note regarding the components of orthopedic implants containing high molecular weight polyethylene (UHMWPE). Rome , March 8, 2005
- Kirkpatrick WN, Jones BM: (2002)The history of Trilucent implants, and a chemical analysis of the triglyceride filler in 51 consecutively removed. Trilucent breast prostheses. J. Plast. Surg, 55:479-89 AND Rizkalla M, Duncan C, Matthews RN. (2001). Trilucent breast implants: a 3 year series. Br J.Plast. Surg. 54: 125-7
- Spear SL, Murphy DK, Silicon A, Walker PS: Inamed Silicone Breast Implant core study results at 6 years. Plast Reconstr Surg. 2007; 120: 8S-165-17S-18S
- Heden P, Nava MB, van Tetering JP, Magalon G, Furie le R, et al. (2006) Prevalence of rupture in inamed silicone breast implants. Plast Reconstr Surg 118:303-8
- Holmich LR, Kjoller K, Vejborg I, Conrad C, Sletting S (2001) Prevalence of silicone breast implant rupture among Danish Women. – Plast. Reconstr Surg 108:848-858
- Parere del Consiglio Superiore di Sanità del 22 Dicembre 2011, Ordinanza Ministero della Salute 29 Dicembre 2011, Legge 5 Giugno 2012 n. 86
- Mongillo R. Vacchiano G (2008) “Medical devices, CE marking of conformity and safeguard measures in light of a Court of Justice recent judgement” Annali Facoltà di Economia di Benevento 13 in press.
- Stefanelli S. Rimondini L: Dispositivi Medici e assicurazione di qualità. Masson Ed. Milano 1999
- Torre M, Romanici E, Calmieri S, Zanali G, Zapponi G (2004) “ Registri degli interventi di protesi d’anca” Not. Ist. Sup. Sanità 17: 3 –10
- Sorenson C, Drummond M (2014) Improving Medical Device Regulation: The United States and Europe in Perspective The Milbank Quarterly 92(1): 114-150
- Cassazione Civile Sez. III Sent. 20 Luglio 1993 n. 8069. Vedi anche Busato A.: I danni da emoderivati: le diverse forme di tutela in Respons. Civ. Prev. 1994, 61
- Petrelli F, Siciliani S, Bernacchia G (2001) Tutela del diritto alla salute da prodotti difettosi e nocivi. Dif. Soc 3:47-60
- Carnevali U (2008) Prodotto difettoso ed oneri probatori del danneggiato. Resp. Civ. Prov. 2:354-359
- Civil Appeal Sec . III Sent. October 8, 2007 n . 20985
- Court of Justice C183 del 4 Agosto 2007, MEDIPAC/ Th Karantzidin A.E. v. Venizelio – Pananio. Case C. 6/05





