Forensic Diagnosis of Acute Myocardial Infarction: Application of Mass Spectrometry
Tsuruyama T1*, Kakimoto Y2
1 Department of Anatomy, Forensic medical and Pathology research, Kyoto University, Kyoto City, Sakyo ku, Yoshida konoe cho 606-8501, Japan.
2 Department of Forensic Medicine, Tokai University, Isehara city, shimokasuya 143, Kanagawa, 259-1193, Japan.
*Corresponding Author
Tatsuaki Tsuruyama MD, PhD,
Department of Anatomy,
Forensic medical and Pathology research,
Kyoto University, Kyoto City,
Sakyo ku, Yoshida konoe cho 606-8501, Japan.
Tel: +81-75-753-4427
E-mail: tsuruyam@kuhp.kyoto-u.ac.jp
Article Type: Review Article
Recieved: September 23, 2014; Accepted: November 10, 2014; Published: November 12, 2014
Citation: Tsuruyama T, Kakimoto Y. (2014), Forensic Diagnosis of Acute Myocardial Infarction: Application of Mass Spectrometry.Int J Forensic Sci Pathol. 2(8), 65-69. doi: dx.doi.org/10.19070/2332-287X-1400018
Copyright: Tsuruyama T© 2014. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
Abstract
Making a precise diagnosis of sudden death due to endogenous acute myocardial infarction in autopsy can be difficult, because histologic myocardial changes and other clinical data are limited. Here, we review previous studies of acute myocardial infarction for pathologic diagnosis using triphenyltetrazolium chloride staining and immunohistochemistry. Further, we introduce our method of forensic examination by mass spectrometry and a novel promising biomarker, SORBS2 for diagnosis of acute lethal cardiac infarction.
2.Introduction
3.Biomarkers for AMI Diagnosis
3.1.Case selection
3.2.Pathology of AMI using TTC
3.3.Immunohistochemistry and TUNEL in diagnosing AMI
3.4.Laser-capture microdissection
3.5.Usability of AMI biomarkers
3.6.SORBS2, a novel promising marker
3.7.Immunostaining of SORBS2 in human heart tissue
4.References
Keywords
Myocardial Infarction; Histologic Observation; Mass Spectrometry; SORBS2.
Introduction
Although acute myocardial infarction (AMI) is one of the most common causes of sudden death outside hospital [1], its molecular pathogenesis is not sufficiently understood. Whereas chronic cardiac infarction lesions show gross fibrosis, and scarring in autopsy, AMI lesions rarely include such changes and it is often difficult to diagnosis on autopsy. For obvious reasons, diagnosis of myocardial infarction by chest pain, or other symptoms such as ST-elevation and/or Q-wave, is limited in autopsy cases, and macroscopic findings are limited and not sufficiently informative at the phase immediately after the onset of the infarction episode. In the autopsied heart, dark mottling is detectable within 4 h after the onset, but varies by case, and a yellow tan may be detectable on damaged tissue within a few days; scarring develops within 8–10 weeks, and is completed by 2 months after the onset. Coronary spasm of an artery in acute coronary syndrome [2] by thrombus formation is so transient that the coronary thrombi are frequently lost in most cases; a guideline for ECG has been recently suggested [3,4].
However, when the AMI is followed by sudden death in the absence of a physician or other trained witness at the scene, forensic diagnosis can be difficult. For this reason, diagnostic findings are guided by histologic examination. In fact, wavy fibers and contraction bands are a hallmark of injured myocardium [5]. However, these injuries take prior to in flammatory neutrophil infiltrates that are also detectable 4–6 h after onset [6]. CXCL8 chemotaxically attracts neutrophils into the myocardium [7]. Neutrophilic infiltrate coronary plaques and infarcted myocardium, and mediate tissue damage by releasing matrix-degrading enzymes and reactive oxygen species. Neutrophils are also involved in adverse postinfarction cardiac remodeling and neointima formation [8]. Thus, neutrophils are pivotal in damaging cardiac myocytes at acute infarction.
Biomarkers for AMI Diagnosis
In the absence of distinct findings such as neutrophil infiltrates, diagnosis of acute infarction of cardiac myocytes can be difficult. For diagnosis of sudden death due to coronary artery disease, ECGs offer essential data; for instance, ST-elevation or saddle back pattern of Q-wave indicate Brugada syndrome, which may cause sudden death [9]. In addition, biochemical data regarding creatinine kinase (CK-MB) is an important diagnostic indicator. In clinical diagnosis, troponins and heart-type fatty acid-binding protein (H-FABP) have been used. Diagnosis by serum markers relies on time-dependent myocardial cell damage that is mainly caused by disruption of coronary artery blood flow (Table 1) (www.j-circ.or.jp/guideline/pdf/JCS2013_kimura_h.pdf). Circulating troponin can be difficult to detect as it may drop to 42% of its concentration at onset within 4 h [10], although more sensitive detection of troponin-T has been investigated [11]. To discover new biomarkers for AMI, gene expression analysis [12] and serum-based proteomics have been useful tools [13]. However, results of blood sample testing at autopsy are often less informative, because of changes during the post-mortem or agonal stages. Another issue is that the above biomarkers are detectable at 4 h after AMI onset, whereas myoglobin and H-FABP can be detected at the early phase of AMI; however, their specificity is lower than that of other markers. Absence of H-FABP staining in the infarcted area and depleted H-FABP staining could be seen in areas that normally show hematoxylin-eosin (H&E) staining [14].
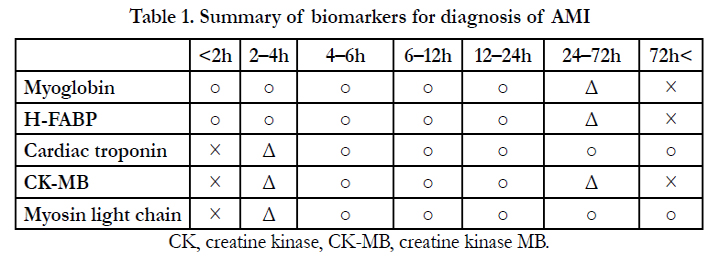

Table 1. Summary of biomarkers for diagnosis of AMI.
CK, creatine kinase, CK-MB, creatine kinase MB.
Although subject selection is critical in identifying AMI biomarkers, it is rather difficult in forensic work because episode onset usually occurs without warning or clinical data. Access to normal controls with no cardiac damage is also essential. We selected tissues from five AMI patients (mean age±SD: 59±12 years) and five normal controls (57±14 years) [15].
Detection of infarcted cardiac myocytes in H&E-stained specimens is often difficult. Sampling cardiac tissues is inevitably accompanied with artifact damage by the sheer stress of cutting a section, and myoctytic direction varies by site in the heart. In the septum, myocyte orientations intersect and run in variable directions. As result, although wavy fibers and contraction bands are a hallmark of infarcted changes, the findings are not necessarily reproducible for pathologists. To prevent such difficulty in histologic observation, longitudinal sections of myocardia are observed microscopically, which shows cardiac linear structure more clearly than do cross-sectioned muscle fibers. Longitudinal stress has been shown to reflect segmental viability in an experimental model [16].
Because we investigated sudden death cases within 12 h after the onset of suspected AMI episodes, inflammatory lesions with neutrophilic infiltration were not evident or not sampled because they would occur a few days after infarction. In the ischemic area of the left ventricle in each AMI heart, we defined two regions: area I contained distinctive contraction bands or wavy fibers, and area II showed other minor irregular patterns, such as fragmented cardiac myocytes. The free wall of the left ventricle was also sampled from each control case. Phospho-tungstic acid-hematoxylin (PTAH) staining was available for accurate detection of damaged cardiac myocytes. As we reported previously [15], PTAH staining has the advantage of showing contraction bands by stainability and contrast of the staining substances. The combination of H&E with PTAH staining was more useful for the detection of cardiac myocytes.
Triphenyltetrazolium chloride (TTC) is a conventional stain for the detection of infarcted areas of the heart in pathological examinations. Myocardialinfarct size was measured by photographs of each gross slice and histologic section [17]. TTC staining shows the activity of mitochondrial dehydrogenase, which is inactivated in myocardial infarction. As the enzyme reduces colorless tetrazolium substance into red formazan (Figure 1), the ischemic lesion is not stained in red. This method was applicable for 12 h in an experimental model using rats that were subjected to coronary ligation.
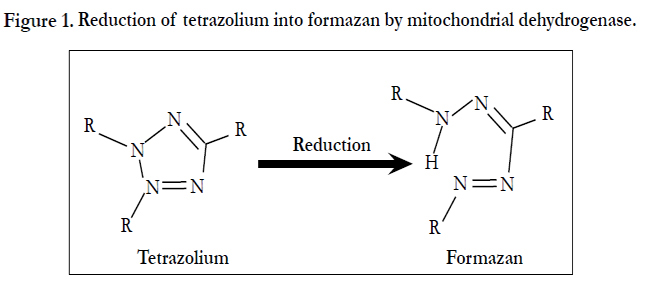
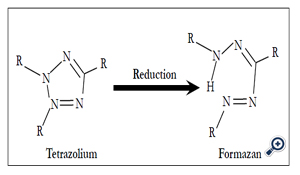
Figure 1. Reduction of tetrazolium into formazan by mitochondrial dehydrogenase.
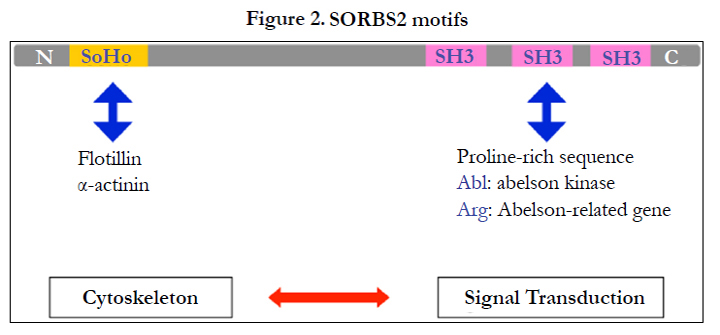
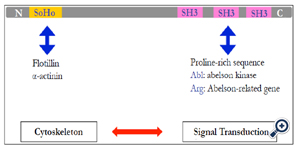
Figure 2. SORBS2 motifs.
We used TTC staining to detect ischemic changes in human autopsies [18]. Although normal tissue did not show definite contrast staining to ischemic lesions, the stainability of TTC declines with the length of the postmortem interval (PMI). Specimens reacted well to TTC within 1.5 days after death and then decreased their stainability logarithmically with PMI (y = (−0.294 Ln [x]) + 1.0441; where x = PMI, y = TTC-stained area/total myocardial area, and R = 0.5673). In older myocardial infarction cases, TTC contrast was clear; but within 9 h after onset, the infarcted area was very difficult to detect [19]. Accordingly, use of TTC to detect AMI in autopsies is relatively limited. Post-mortem degeneration of mitochondrial protein, including dehydrogenase, is probably inevitable.
Immunohistochemistry has also been used to diagnose AMI. Antibodies to fibronectin [20], troponin I [21], cardiac myosinbinding protein C [22,23], and IL-9 [24] have been used to detect injured cardiac myocytes, and antibody to von Willebrand factor has been applied to lung tissue to find the cause of death [25]. Transferase-mediated deoxyuridine triphosphate nick end-labeling (TUNEL) staining for apoptosis is also used to detect injured cardiac myocytes [26]. Although no difference was seen between patients with and without coronary artery disease (P>0.05), TUNEL is useful in cases where sudden cardiac death is suspected; however, we found no specificity for acute ischemic changes in our study; TUNEL staining showed similar results (data not shown). Although troponin-T was also available to detect injured cardiac myocytes [21]; their stainability was not reproducible in the use of autopsy samples. In particular, this method is not confirmative in lesions with low stainability, because low troponin stainability does not indicate AMI pathogenesis. To distinguish lipofuschin deposits in cardiac myocytes, we used permanent red as a chromogen instead of routine 3,3'-diaminobenzidine [27] because cardiac myocytes in subjects older than middle-aged contain abundant lipofuscin, a brown pigment that is easily confused with 3,3'-diaminobenzidine. We excluded large capillaries and fibrotic areas. Envision System/AP, Rabbit/Mouse (Dako product, Agilent Technology, Santa Clara, CA) was used for detection [15].
Generally, formaldehyde-fixed and paraffin-embedded specimens have been considered inferior to frozen sections in protein analysis. However, we applied the following protocol that was reported by us [28] and has proved to be sufficient. Laser caption microdis-section was performed using an LMD 6500 (Leica Microsystems, Wetzlar, Germany). Cardiac tissues of 10 mm2 in total were collected into a 500-μL well. Large capillaries and areas of fibrosis were excluded to prevent contaminating other types of injury. To extract proteins followed by liquid chromatography/mass spectrometry (LC/MS), each microdissected sample was suspended in 20 μL of 0.1 mol/L NH4HCO3 containing 30% (v/v) CH3CN and centrifuged at 10,000g for 1 min. Tubes were heated at 95° C for 90 min. After retrieval, centrifugation, and cooling, trypsin digestion was performed. Tubes were incubated at 37°C overnight, and then heated at 95°C for 5 min to deactivate the trypsin. After drying, samples were resuspended in 0.1% trifluoroacetic acid with 2% CH3CN, resulting in a final protein concentration of 0.2 μg/μL.
To our experience, 1.0×104 peptides and 102–3 proteins could be identified, with a false-discovery rate of < 1% in the use of various formaldehyde fixed paraffin embedded (FFPE)tissues including heart, kidney, pancreas, and brain, in numbers not necessarily inferior to frozen samples. Indeed, a previous study reported that frozen-sample proteins rapidly degenerate in 2 years, even when kept at ≤−80°C. To obtain sensitive and specific markers, non-specific abundant proteins should be excluded systematically. Collagen, keratin, and other cytoskeletal proteins including beta-tubulin and vimentin are such proteins (Table 2). In addition, immunoglobulin light chain, serum albumin, and hemoglobin-β chain are frequently detectable in MS of both FFPE and frozen tissues. Our previous studies found levels of 21 proteins to differ significantly between AMI patients and control subjects. According to Gene Ontology (www.geneontology.org/) analysis, cytoplasmic, mitochondrial, and extramyocardial proteins were detected, as validated by detection of the conventional marker, H-FABP.
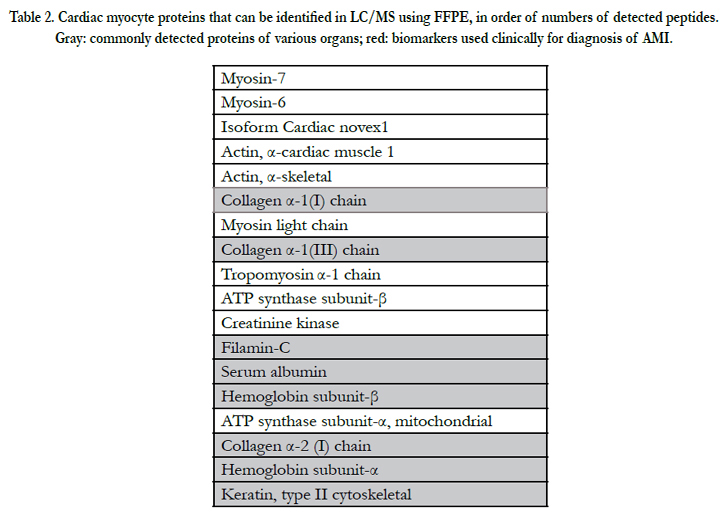
Table 2.Cardiac myocyte proteins that can be identified in LC/MS using FFPE, in order of numbers of detected peptides.Gray: commonly detected proteins of various organs.
Gray: commonly detected proteins of various organs; red: biomarkers used clinically for diagnosis of AMI.
The feasibility of using identified proteins is limited in clinical examination. Sarcomeric protein is considered a diagnostic biomarker because it is specific to cardiac muscle. Similarly, serum troponin is thought to be always detectable. H-FABP, an established clinical diagnostic marker, is detectable within the 4 h after the onset of clinical symptoms. However, the stability of proteins in the postmortem period is an important limitation. After sudden death, autolysis or digestion rapidly progresses, and most proteins degenerate. For this reason, utility of the markers in Table 1 is limited by time. In our previous study, H-FABP, myosin-binding protein, and cardiac troponin I were detectable [15]; however, some proteins are not stable and western blot failed to detect them.
Using LC/MS, we found the marker Sorbin and SH3 domain-containing protein-2, (SORBS2) [16]. Another signature is ARGBP2, which interacts with Arg–ABL binding proteins via SH3 domains at its carboxyl end (Figure 2) [29-32]. This protein carries 3 Cterminal SH3 domains [33] and an N-terminal sorbin homology (SoHo) domain that interact with lipid-raft proteins. Abelson kinase (ABL) may interact with SH3; flotillin and α-actinin interact at the SoHo domain. The two functional domains let SOBS2 mediate cytoskeletal change by physical stress and signal transduction through ABL. This protein is encoded by proto-oncogenes that recombine with the BCR gene in the Philadelphia chromosome derived from t(9;22) in patients with chronic myeloid leukemia.
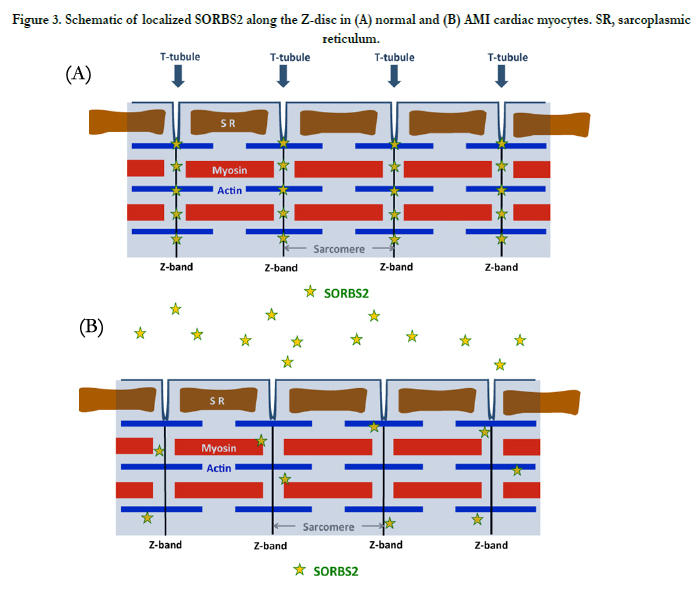
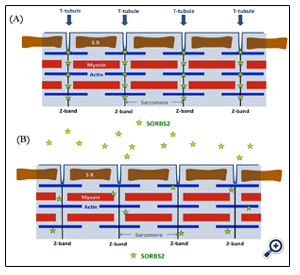
Figure 3. Schematic of localized SORBS2 along the Z-disc in (A) normal and (B) AMI cardiac myocytes. SR, sarcoplasmic reticulum.
SORBS2 normally localizes along the Z-line in myocardial cytoplasm and is observed in the intercalated disk in control heart tissue. In AMI tissue, we have observed that SORB2 alignment in the Z-bands is disordered at the contraction bands. The schematic in Figure 3 shows the localized SORBS2 along the Z-line and its release in AMI. By leaking SORBS2, cardiac muscle may lose the response to physical stress and fragmentation may develop. Notably, as we reported previously, this protein can be maintained even a week after death in peripheral blood using western blot analysis and is a forensic medical marker. We found using western blot analysis that SORBS2 levels are marked higher in serum of AMI patient than in control serum. These observations indicate that SORBS2 is released from cardiac myocytes in an acute lethal infarction.
Here, we introduced a novel method for extracting proteins from FFPE tissue that enables proteomic analysis. Label-free LC-MS that uses precise microscopic sampling successfully identified a biomarker for forensic diagnosis. Although further clinical research using AMI serum is necessary to clarify the usefulness of SORBS2 in early diagnosis, this sarcoplasmic protein could help assess ischemic severity in cardiac tissue.
References
- Adnet F, Renault R, Jabre P, Kulstad E, Galinski M, et al. (2011) Incidence of acute myocardial infarction resulting in sudden death outside the hospital. Emerg Med J 28: 884-886.
- Elhofy A, Depaolo RW, Lira SA, Lukacs NW, Karpus WJ (2009) Mice deficient for CCR6 fail to control chronic experimental autoimmune encephalomyelitis. J Neuroimmunol 213: 91-99.
- Muller C (2012) New ESC guidelines for the management of acute coronary syndromes in patients presenting without persistent ST-segment elevation. Swiss Med Wkly 142: w13514.
- Hamm CW, Bassand JP, Agewall S, Bax J, Boersma E, et al. (2011) ESC Guidelines for the management of acute coronary syndromes in patients presenting without persistent ST-segment elevation: The Task Force for the management of acute coronary syndromes (ACS) in patients presenting without persistent ST-segment elevation of the European Society of Cardiology (ESC). Eur Heart J 32: 2999-3054.
- Nakatani S, McCarthy PM, Kottke-Marchant K, Harasaki H, James KB, et al. (1996) Left ventricular echocardiographic and histologic changes: impact of chronic unloading by an implantable ventricular assist device. J Am Coll Cardiol 27: 894-901.
- Steiner I, Spacek J, Matejkova A, Vojacek J, Bis J, et al. (2014) Histopathology of aspirated thrombi during primary percutaneous coronary intervention in patients with acute myocardial infarction. Cardiovasc Pathol 23: 267-271.
- Dyer DP, Thomson JM, Hermant A, Jowitt TA, Handel TM, et al. (2014) TSG-6 inhibits neutrophil migration via direct interaction with the chemokine CXCL8. J Immunol 192: 2177-2185.
- Carbone F, Nencioni A, Mach F, Vuilleumier N, Montecucco F (2013) Pathophysiological role of neutrophils in acute myocardial infarction. Thromb Haemost 110: 501-514.
- Antzelevitch C, Brugada P, Borggrefe M, Brugada J, Brugada R, et al. (2005) Brugada syndrome: report of the second consensus conference: endorsed by the Heart Rhythm Society and the European Heart Rhythm Association. Circulation 111: 659-670.
- Haltern G, Peiniger S, Bufe A, Reiss G, Gulker H, et al. (2010) Comparison of usefulness of heart-type fatty acid binding protein versus cardiac troponin T for diagnosis of acute myocardial infarction. Am J Cardiol 105: 1-9.
- Marini MG, Cardillo MT, Caroli A, Sonnino C, Biasucci LM (2013) Increasing specificity of high-sensitivity troponin: new approaches and perspectives in the diagnosis of acute coronary syndromes. J Cardiol 62: 205-209.
- Xu F, Teng X, Yuan X, Sun J, Wu H, et al. (2014) LCK: a new biomarker candidate for the early diagnosis of acute myocardial infarction. Mol Biol Rep.
- Aydin S, Aydin S, Kobat MA, Kalayci M, Eren MN, et al. (2014) Decreased saliva/serum irisin concentrations in the acute myocardial infarction promising for being a new candidate biomarker for diagnosis of this pathology. Peptides 56: 141-145.
- Meng X, Ming M, Wang E (2006) Heart fatty acid binding protein as a marker for postmortem detection of early myocardial damage. Forensic Sci Int 160: 11-16.
- Kakimoto Y, Ito S, Abiru H, Kotani H, Ozeki M, et al. (2013) Sorbin and SH3 domain-containing protein 2 is released from infarcted heart in the very early phase: proteomic analysis of cardiac tissues from patients. J Am Heart Assoc 2: e000565.
- Aarsaether E, Rosner A, Straumbotn E, Busund R (2012) Peak longitudinal strain most accurately reflects myocardial segmental viability following acute myocardial infarction - an experimental study in open-chest pigs. Cardiovasc Ultrasound 10: 23.
- Fishbein MC, Meerbaum S, Rit J, Lando U, Kanmatsuse K, et al. (1981) Early phase acute myocardial infarct size quantification: validation of the triphenyl tetrazolium chloride tissue enzyme staining technique. Am Heart J 101: 593-600.
- Adegboyega PA, Adesokan A, Haque AK, Boor PJ (1997) Sensitivity and specificity of triphenyl tetrazolium chloride in the gross diagnosis of acute myocardial infarcts. Arch Pathol Lab Med 121: 1063-1068.
- Kakimoto Y, Tsuruyama T, Miyao M, Abiru H, Sumiyoshi S, et al. (2013) The effectiveness and limitations of triphenyltetrazolium chloride to detect acute myocardial infarction at forensic autopsy. Am J Forensic Med Pathol 34: 242-247.
- Hu BJ, Chen YC, Zhu JZ (1996) Immunohistochemical study of fibronectin for postmortem diagnosis of early myocardial infarction. Forensic Sci Int 78: 209-217.
- Hansen SH, Rossen K (1999) Evaluation of cardiac troponin I immunoreaction in autopsy hearts: a possible marker of early myocardial infarction. Forensic Sci Int 99: 189-196.
- Jacquet S, Yin X, Sicard P, Clark J, Kanaganayagam GS, et al. (2009) Identification of cardiac myosin-binding protein C as a candidate biomarker of myocardial infarction by proteomics analysis. Mol Cell Proteomics 8: 2687-2699.
- Govindan S, McElligott A, Muthusamy S, Nair N, Barefield D, et al. (2012) Cardiac myosin binding protein-C is a potential diagnostic biomarker for myocardial infarction. J Mol Cell Cardiol 52: 154-164.
- Gregersen I, Skjelland M, Holm S, Holven KB, Krogh-Sorensen K, et al. (2013) Increased systemic and local interleukin 9 levels in patients with carotid and coronary atherosclerosis. PLoS One 8: e72769.
- Quan L, Ishikawa T, Zhao D, Michiue T, Yoshida C, et al. (2009) Immunohistochemistry of von Willebrand factor in the lungs with regard to the cause of death in forensic autopsy. Leg Med (Tokyo) 11 Suppl 1: S294-296.
- Edston E, Grontoft L, Johnsson J (2002) TUNEL: a useful screening method in sudden cardiac death. Int J Legal Med 116: 22-26.
- Assefnia S, Kang K, Groeneveld S, Yamaji D, Dabydeen S, et al. (2014) Trp63 is regulated by STAT5 in mammary tissue and subject to differentiation in cancer. Endocr Relat Cancer 21: 443-457.
- Kakimoto Y, Tsuruyama T, Yamamoto T, Furuta M, Kotani H, et al. (2012) Novel in situ pretreatment method for significantly enhancing the signal in MALDI-TOF MS of formalin-fixed paraffin-embedded tissue sections. PLoS One 7: e41607.
- Pession A, Lo Nigro L, Montemurro L, Serravalle S, Fazzina R, et al. (2006) ArgBP2, encoding a negative regulator of ABL, is fused to MLL in a case of infant M5 acute myeloid leukemia involving 4q35 and 11q23. Leukemia 20: 1310-1313.
- Hand D, Eiden LE (2005) Human sorbin is generated via splicing of an alternative transcript from the ArgBP2 gene locus. Peptides 26: 1278-1282.
- Cestra G, Toomre D, Chang S, De Camilli P (2005) The Abl/Arg substrate ArgBP2/nArgBP2 coordinates the function of multiple regulatory mechanisms converging on the actin cytoskeleton. Proc Natl Acad Sci U S A 102:1731-1736.
- Wang B, Golemis EA, Kruh GD (1997) ArgBP2, a multiple Src homology 3 domain-containing, Arg/Abl-interacting protein, is phosphorylated in v- Abl-transformed cells and localized in stress fibers and cardiocyte Z-disks. J Biol Chem 272: 17542-17550.
- Roignot J, Bonacci T, Ghigo E, Iovanna JL, Soubeyran P (2014) Oligomerization and phosphorylation dependent regulation of ArgBP2 adaptive capabilities and associated functions. PLoS One 9: e87130.





