Obesity, Diabetes and Cancer: A Mechanistic Perspective
Cifarelli V1*, Hursting SD2,3
1 Department of Medicine, Center for Human Nutrition, Washington University School of Medicine, St. Louis, MO, USA.
2 Department of Nutrition, University of North Carolina, Chapel Hill, NC, USA.
3 Lineberger Comprehensive Cancer Center, University of North Carolina, Chapel Hill, NC, USA.
*Corresponding Author
Vincenza Cifarelli,
Department of Medicine, Center for Human Nutrition,
Washington University School of Medicine, St. Louis, MO 63110, USA.
Tel: (314) 362 8206
Fax: (314) 362 8230
E-mail: vcifarel@dom.wustl.edu
Received: July 24, 2015; Accepted: September 25, 2015; Published: September 28, 2015
Citation: Cifarelli V, Hursting SD (2015) Obesity, Diabetes and Cancer: A Mechanistic Perspective. Int J Diabetol Vasc Dis Res, S4:001, 1-7. doi: dx.doi.org/10.19070/2328-353X-SI04001.
Copyright: Cifarelli V© 2015. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
Nearly 35% of adults and 20% of children in the United States are obese, defined as having a body mass index (BMI) ≥ 30 kg/m2. Obesity is an established risk factor for many cancers, and obesity-associated metabolic perturbations often manifest in Type 2 diabetes mellitus and/or the metabolic syndrome. As part of the growth-promoting, proinflammatory microenvironment of the obese and/or diabetic state, crosstalk between macrophages, adipocytes, and epithelial cells occurs via metabolically-regulated hormones, cytokines, and other mediators to enhance cancer risk and/or progression. This review synthesizes the evidence on key biological mechanisms underlying the associations between obesity, diabetes and cancer, with particular emphasis on enhancements in growth factor signaling, inflammation, and vascular integrity processes. These interrelated pathways represent mechanistic targets for disrupting the obesity-diabetes-cancer link, and several diabetes drugs, such as metformin and rosiglitazone, are being intensely studied for repurposing as cancer chemopreventive agents.
2.Introduction
3.Dysregulated Growth Signals
3.1 Insulin and IGF-I
3.2 Signaling Pathways Downstream of the Insulin Receptor and IGF-1R
3.3 Leptin, Adiponectin and their Ratio
4.Chronic Inflammation
4.1 Cytokines and Crosstalk Between Adipocytes, Macrophages and Epithelial Cells
4.2 Inflammation and Cancer
5.Vascular Integrity-Related Factors
5.1 Plasminogen Activator Inhibitor-1 (PAI-1)
5.2 Vascular Endothelial Growth Factor (VEGF)
5.3 Tumor angiogenesis
6.Cancer and Diabetes: Role of Antioxidant
7.Cancer and Antidiabetic Drugs
7.1 Metformin
7.2 Rosiglitazone
8.Conclusion
9.Acknowledgements
10.References
Keywords
Obesity; Diabetes; Cancer; Metformin.
Introduction
The prevalence of obesity, defined as having a body mass index (BMI) ≥30 kg/m2, has increased dramatically in recent decades in the United States, and more than 35% of adults and 20% of children are now obese [1]. Among obese individuals, approximately 65% of males and 56% of females meet the criteria for the metabolic syndrome, a state of metabolic dysregulation characterized by insulin resistance, hyperglycemia, dyslipidemias (particularly hypertriglyceridemia), and hypertension [2]. In obesity and/or metabolic syndrome, alterations also occur in circulating levels of insulin, bioavailable insulin-like growth factor (IGF)-1, adipokines (eg, leptin and adiponectin), inflammatory factors (eg, cytokines), and vascular integrity-related factors (eg, plasminogen activator inhibitor [PAI]-1 and vascular endothelial growth factor [VEGF]) [3, 4]. Through these mediators, and likely several others, obesity and metabolic syndrome are linked to various chronic diseases [3,5] including cardiovascular disease, Type II diabetes (T2D), and the focus of this review, cancer.
Evidence-based guidelines for cancer prevention urge avoiding behaviors that can lead to obesity, including overconsumption of energy-dense foods and beverages and a sedentary lifestyle [6]. Overall, 20-25% of all cancer deaths in the US have been attributed to overweight and obesity [7]. In their position statement on obesity and cancer, the American Society of Clinical Oncology states that obesity has emerged as the leading preventable cause of cancer that can also increase risk of cancer recurrence and lower survival. They predict that the US will see 500,000 new obesity- related cancer cases per year by 2030 unless corrective action is taken [8]. In terms of specific cancers, obesity is associated with increased mortality from cancer of the prostate and stomach in men; breast (postmenopausal), endometrium, cervix, uterus and ovaries in women; and kidney (renal cell), colon, esophagus (adenocarcinoma), stomach, pancreas, gallbladder, and liver in both genders [9].
T2D is a progressive metabolic disorder associated with overweight, obesity and several pathological abnormalities including glucose intolerance, insulin resistance, and dyslipidemia. In the United States, 1.9 million of people are newly diagnosed with diabetes mellitus and 90%-95% of all diagnosed are affected by T2D [10]. Emerging evidence indicates that T2D associates with an increased risk of cancer at specific sites. The risk of developing pancreatic, hepatocellular, or endometrial cancer is almost twice in diabetic patients compared to that of the nondiabetic population [10-15]. Other types of cancer that associate with diabetes are breast [16], bladder [17, 18], kidney [19], and colorectal cancer [20]. No association has been identified between diabetes and prostate cancer [21, 22]. It is still not understood why diabetes associates with increased risk of developing some but not all types of cancers, and further investigation is needed regarding this issue.
Herein, we discuss possible mechanisms underlying the links between obesity, T2D and cancer, with emphasis on obesity-associated enhancements in growth factor signaling, inflammation, and angiogenic processes and on the crosstalk between macrophages, adipocytes, endothelial cells and epithelial cells in many cancers.
Dysregulated Growth Signals
Insulin is a peptide hormone produced by pancreatic beta cells and released in response to elevated blood glucose. Hyperglycemia, a hallmark of metabolic syndrome, is associated with insulin resistance, aberrant glucose metabolism, chronic inflammation, and the production of other metabolic hormones such as IGF-I, leptin and adiponectin [23]. Sharing ~50% sequence homology with insulin, IGF-I is a peptide growth factor produced primarily by the liver following stimulation by growth hormone. IGF-I regulates growth and development of many tissues, particularly prenatally [24]. IGF-I in circulation is typically bound to IGF binding proteins (IGFBPs) that regulate the amount of free IGF-I bioavailable to bind to the IGF-I receptor (IGF-IR) and elicit growth or survival signaling [24]. In metabolic syndrome, the amount of bioavailable IGF-I increases, possibly via hyperglycemia-induced suppression of IGFBP synthesis and/or hyperinsulinemia-induced promotion of hepatic growth hormone receptor expression and IGF-I synthesis [23]. Elevated circulating IGF-I is an established risk factor for many cancer types [25].
In many tumors, binding of IGF-I and IGF-II to the IGF-IR inhibits apoptosis and promotes cell proliferation. IGF-I can also bind and form heterodimers with insulin receptors subtypes A and B (IR-A and IR-B), generating hybrids. IR-A has high affinity for insulin and IGF-II but binds IGF-I with low affinity. IR-A binds insulin with 1.5-fold higher affinity than IR-B, possesses a higher dissociation and internalization rate, and mimics many of the cell signaling and cell survival activities of IGF-IR (described below). In contrast, IR-B is thought to mediate most of the classic metabolic responses induced by insulin, and also binds IGF-I and IGF-II with low and intermediate affinity, respectively [26]. Both IGFs- and insulin-mediated induction of cell proliferation and migration is more effective in cells containing IGF-IR/IR-A hybrids than in cells containing IGF-IR/IR-B hybrids [27]. Interestingly, an increase in the IR-A to IR-B ratio has been reported in Type 2 diabetes [28, 29]. Activation of the IR-A by insulin-like IGF-II bypasses the IGF-IR and its inhibition may explain the reason of the failure of monotherapy trials with IGF-IR targeted antibody or with IGF-IR specific tyrosine kinase inhibitors [30].
Downstream targets of IR-A and IGF-IR comprise a signaling network regulating cellular growth and survival predominately through induction of the phosphatidylinositol-3 kinase (PI3K)/ Akt pathway [24]. This signaling cascade, elegantly reviewed by Cantley and colleagues [31], is one of the most commonly altered pathways in human epithelial tumors. Engagement of the PI3K/Akt pathway allows both intracellular and environmental cues, such as energy availability and growth factor supply, to affect cell growth, proliferation, survival and metabolism. Activation of the insulin receptor or IGF-IR stimulates PI3K to produce the lipid second messenger, phosphatidyl-inositol-3,4,5-trisphosphate. This messenger recruits and anchors Akt to the cell membrane for further phosphorylation and activation. Akt is a cAMP-dependent, cGMP-dependent protein kinase C that when constitutively active is sufficient for cellular transformation by stimulating cell cycle progression and cell survival and by inhibiting apoptosis. Frequently associated with the aberrant Akt signaling commonly seen in human cancers is an increase in mTOR (mammalian target of rapamycin) signaling [32]. mTOR is a serine/threonine protein kinase that is activated by Akt and inhibited by an opposing signal from AMP-activated kinase (AMPK). At the interface of the Akt and AMPK pathways, mTOR dictates translational control of new proteins in response to both growth factor signals and nutrient availability through phosphorylation of its downstream mediators [33, 34]. Ultimately, activation of mTOR results in cell growth, cell proliferation, and resistance to apoptosis. Additionally, elevated cellular amino acids (particularly branched chain amino acids), glucose, and ATP concentrations, which occur during high-energy conditions, signal for mTOR activation [34].
Leptin is a peptide hormone produced by adipocytes, is positively correlated with adipose stores and nutritional status, and functions as an energy sensor to signal the brain to reduce appetite. In the obese state, adipose tissue overproduces leptin, and the brain no longer responds to the signal. Insulin, glucocorticoids, tumor necrosis factor-alpha (TNF-α), and estrogens all stimulate leptin release [35, 36]. Leptin has direct effects on peripheral tissues, indirect effects on hypothalamic pathways and modulates immune function, cytokine production, angiogenesis, carcinogenesis and other biological processes [36]. The leptin receptor has similar homology to class I cytokines that signal through the janus kinase and signal transducer activator of transcription (JAK/STAT) pathway that is often dysregulated in cancer [36].
Adiponectin is a hormone mainly secreted from visceral adipose tissue. Levels of adiponectin, in contrast with leptin, negatively correlate with adiposity. Adiponectin functions to counter the metabolic program associated with obesity and hyperleptinemia by modulating glucose metabolism, increasing fatty acid oxidation and insulin sensitivity, and decreasing production of inflammatory cytokines [37]. The possible mechanisms through which adiponectin exerts anticancer effects may include increasing insulin sensitivity, and decreasing insulin/IGF-1 and mTOR signaling via activation of AMPK. Adiponectin also reduces (and leptin increases) proinflammatory cytokine expression via modulation of JAK/STAT pathway and the the nuclear factor kappa-lightchain- enhancer of activated B-cells (NF-kB) [38-40]. Although in vitro, animal and epidemiologic evidence linking leptin [41-44] or adiponectin [39-45] individually to cancer risk is mixed, data suggest that the adiponectin:leptin ratio is a useful atherogenic and metabolic syndrome index, particularly in Type 2 diabetics, and is also associated with cancer risk [45]. However, further characterization of these links is needed.
Chronic Inflammation
Obesity and metabolic syndrome are associated with a low-grade, chronic state of inflammation characterized by increased circulating free fatty acids and chemoattraction of immune cells (such as macrophages that also produce inflammatory mediators) into the local milieu [46-48]. These effects are further amplified by the release of inflammatory cytokines such as interleukin (IL)-1β, IL- 6, TNF α and monocyte chemoattractant protein (MCP)-1. Adipocytes can enlarge past the point of effective oxygen diffusion, which results in hypoxia and eventually necrosis. Free fatty acids escape the engorged/necrotic adipocytes and deposit in other tissues, which in turn promotes insulin resistance and diabetes (through downregulation of insulin receptors and glucose transporters), hypertension, and fatty liver disease and also activates signaling molecules involved in epithelial carcinogenesis, such as NF-kB [49, 50].
NF-кB is a transcription factor that is activated in response to bacterial and viral stimuli, growth factors, and inflammatory molecules (eg, TNF-α, IL-6, and IL-1β), and is responsible for inducing gene expression associated with cell proliferation, apoptosis, inflammation, metastasis, and angiogenesis. Activation of NF-кB is a common characteristic of many tumors and is associated with insulin resistance and elevated circulating levels of leptin, insulin, and/or IGF-1 [49, 50].
The link between chronic inflammation and cancer development was first noticed more than 100 years ago by Rudolph Virchow when he observed an abundance of leukocytes in neoplastic tissue [51]. Now, several tissue-specific inflammatory lesions are established neoplastic precursors for invasive cancer, including gastritis for gastric cancer, inflammatory bowel disease for colon cancer, and pancreatitis for pancreatic cancer [52, 53].
Tumor microenvironments are composed of multiple cell types including epithelial cells, fibroblasts, mast cells, and cells of the innate and adaptive immune system [53, 54]. As discussed previously, macrophages, which are activated in the obese state, infiltrate tumors and amplify the inflammatory tumor microenvironment through production of cytokines, prostaglandins, and angiogenic factors [53]. Another important cancer-related inflammatory mediator is cyclooxygenase (COX)-2, an enzyme that is upregulated in most tumors and catalyzes the synthesis of the potent inflammatory lipid metabolite, prostaglandin E2. COX-2 overexpression is an indicator of poor prognosis in multiple cancer types [55].
Vascular Integrity-Related Factors
PAI-1 is a serine protease inhibitor produced by endothelial cells, stromal cells and adipocytes in visceral white adipose tissue [62]. Increased circulating PAI-1 levels, frequently found in obese subjects, are associated with increased risk of atherogenesis and cardiovascular disease, diabetes and several cancers [4, 56]. PAI-1, through its inhibition of urokinase-type and tissue-type plasminogen activators, regulates fibrinolysis and integrity of the extracellular matrix. PAI-1 is also involved in angiogenesis and thus may contribute to obesity-driven tumor cell growth, invasion and metastasis [4].
VEGF, a heparin-binding glycoprotein produced by adipocytes and tumor cells, has angiogenic, mitogenic and vascular permeability- enhancing activities specific for endothelial cells [57]. Circulating levels of VEGF are increased in obese, relative to lean, humans and animals, and increased tumoral expression of VEGF is associated with poor prognosis in several obesity-related cancers [58]. The need for nutrients and oxygen triggers tumor cells to produce VEGF, which leads to the formation of new blood vessels to nourish the rapidly growing tumor and facilitate the metastatic spread of tumors cells [57]. Adipocytes communicate with endothelial cells by producing a variety of proangiogenic and vascular permeability-enhancing factors. These include VEGF, IGF-1, PAI-1, leptin, hepatocyte growth factor, and fibroblast growth factor-2 [59]. In the obese, nontumor setting, these factors stimulate neovascularization in support of the expanding fat mass. These adipose-derived factors may also contribute to obesity- associated enhancement of tumor angiogenesis. However, the relative contributions of tumor-derived, versus adipocyte-derived, proangiogenic factors in tumor development, progression and metastasis remain unclear.
Adipocytes communicate with endothelial cells by producing a variety of proangiogenic and vascular permeability-enhancing factors, including vascular endothelial growth factor (VEGF), IGF-1, plasminogen activator inhibitor (PAI)-1, leptin, hepatocyte growth factor, and fibroblast growth factor-2 [59]. In the obese state, these factors stimulate neovascularization in support of the expanding fat mass. Insulin resistance also drives abornomalities in endothelial and vascular smooth muscle function via alterations in many of the same proangiogenic and vasular altering factors [59]. These factors may also contribute to obesity-associated enhancement of tumor angiogenesis. For example, VEGF is a heparin-binding glycoprotein produced by adipocytes and tumor cells that has angiogenic, mitogenic and vascular permeabilityenhancing activities specific for endothelial cells [57]. Circulating levels of VEGF are increased in obese, relative to lean, humans and animals, and increased tumoral expression of VEGF is associated with poor prognosis in several obesity-related cancers [58]. The need for nutrients and oxygen triggers tumor cells to produce VEGF, which leads to the formation of new blood vessels to nourish the rapidly growing tumor and may facilitate the metastatic spread of tumors cells [57]. Bevacizumab-based therapy (ie, anti- VEGF therapy), in combination with conventional chemotherapy, is considered a first-line treatment option for patients with advanced colorectal cancer; however, decreased efficacy in obese patients is reported and speculated to be associated with increased levels of VEGF (and/or other proangiogenic factors) produced by visceral white adipose tissue [60, 61]. The relative contributions of tumor-derived, versus adipocyte-derived, proangiogenic factors in tumor development, progression and metastasis remain unclear. PAI-1 is a serine protease inhibitor produced by endothelial cells, stromal cells and adipocytes in visceral white adipose tissue [56]. Increased circulating PAI-1 levels, frequently found in obese subjects, are associated with increased risk of atherogenesis and cardiovascular disease, diabetes and several cancers [56, 4]. PAI-1, through its inhibition of urokinase-type and tissue-type plasminogen activators, regulates fibrinolysis and integrity of the extracellular matrix. PAI-1 is also involved in angiogenesis and thus may contribute to obesity-driven tumor cell growth, invasion and metastasis [4].
Cancer and Diabetes: Role of Antioxidant
T2D shares multiple risk factors with several cancers, such as obesity, hyperinsulinemia and hyperglycemia. The cancer-obesity association has been known for many years [62, 63], and could be explained by the fact that central obesity often leads to chronic levels of inflammation and oxidative stress [64], which are risk factors for the development of many cancers. Excess body weight and adiposity associates with reduced insulin sensitivity, which is compensated by increased insulin secretion, to avoid an increase in blood glucose level [65]. Obesity-related hyperinsulinemia regulates the secretion of adipokines by adipocytes that together with macrophages infiltrating the adipose tissue, secrete a number of pro-inflammatory cytokines as tumor necrosis factor (TNF)-α and interleukin-(IL)-6. The induced systemic inflammatory response worsens insulin resistance [66], contributing to increase the risk of pancreatic cancer through insulin/IGF-1 signaling cascades.
Obesity-induced reactive oxygen species (ROS) production represents an additional risk factor for the development of pancreatic cancer in T2D patients. Hyperglycemia increases oxidative stress and up-regulate the oxidative machinery. Specifically, hyperglycemia activates the polyol pathway through increased expression and activity of aldose reductase. It has been hypothesized that the hyperglycemia-induced activation of polyol metabolism may explain the decreased pancreatic secretion observed in insulindependent diabetes mellitus patients [67, 68]. In vitro, glucoseinduced activation of sorbitol accumulation and nuclear factor- κB (NFκB) activation can be prevented by normalizing levels of mitochondrial ROS [69]. Additional studies have also reported lower levels of other cellular antioxidants, namely copper/zinc superoxide dismutase, catalase, and glutathione peroxidase (GPx), in human pancreatic cancer specimens compared with normal pancreatic specimens [67, 70]. Although these mechanisms still need to be more clarified, these findings suggest that cellular antioxidants play a crucial role in regulating pancreatic tumor growth.
Cancer and Antidiabetic Drugs
Metformin (1,1-dimethylbiguanide hydrocloride) is a member of the biguanide class of oral agents developed for the treatment of hyperglycaemia and T2D. Discovered more than 50 years ago, metformin still represents the most commonly prescribed drug for T2D patients as initial or combined therapy [71]. Epidemiologic studies suggest that metformin use is associated with reduced risk among diabetics of being either diagnosed with, or dying from several cancers, particularly pancreatic cancer (PC) [72]. However, the causal relationship and potential mechanisms underlying the association between metformin and reduced PC have not been established.
With regard to diabetes, metformin decreases hyperglycemia by inhibiting hepatic gluconeogenesis and increasing glucose uptake in the peripheral tissues, as skeletal muscles and adipose tissue, ultimately improving insulin sensitivity [73]. These effects are achieved by activation of AMPK a sensor of the cellular metabolic state [74, 75]. Metformin-induced AMPK negatively regulates mTOR pathway, which is involved in protein synthesis and cell proliferation. With respect of cancer, metformin might directly achieve inhibition of tumor cell growth through induction of AMPK. Indirectly, metformin affects tumor growth in T2D patients by decreasing insulin, a promoting-growth hormone, with mitogenic effects. It has been proposed that hyperinsulinemia together with insulin resistance might promote carcinogenesis [76] since several in vivo studies have shown that antitumour effect of metformin is more effective in mice on high-energy diet associated with hyperinsulinemia than those on control diet [77]. Metformin has been shown to inhibit cell proliferation, colony formation and partial cell cycle arrest [78-80] in several cancer cell lines. In addition to the above mechanisms, metformin may also exert its anti-tumor effect through regulation of microRNA (miRNAs), noncoding RNA molecules that post-transcriptionally regulate gene expression [81]. MiRs regulate cellular processes involved in cancer initiation, recurrence, and metastasis [82]. Our recent study conducted in a murine pancreatic cancer transplant model in obese/prediabetic mice found that metformin induced tumoral expression of miRNA-34a [83], a miR typically lost during cancer progression, including pancreatic tumors [84, 85]. MiR-34a has been shown to contribute to reduce survivability in those diagnosed with pancreatic cancer [86]. MicroRNA-34a suppresses key events of the epithelial-to-mesenchymal transition (EMT) by acting as a negative regulator of the signaling pathway activated by transforming growth factor-β (TGF-β), a master regulator of EMT [87]. Expression of EMT mediators, such as Snail is profoundly linked to TGF-β signaling and miR-34a expression. Specifically, as TGF-β increases, expression of miR-34a is diminished and Snail is enhance [88], while re-expression of miR-34a reduces expression of Snail and other EMT mediators such as Notch and Slug [89]. Metformin modulates EMT-related targets, such as Notch, in a miR-34a-dependent manner [90]. Dysregulation of Notch signaling, a targeted perturbation in many cancer types has been shown to enhance hypoxia-induced tumor cell migration and invasion and influence expression of Snail and Slug [91].
Rosiglitazone is a thiazolidinedione that reduces insulin resistance and might preserve insulin secretion. The thiazolidinediones are agonists for peroxisome-proliferator–activated receptor γ (PPAR-γ). PPAR-γ receptors are ligand-activated nuclear transcription factors that modulate gene expression, lowering blood glucose primarily by increasing insulin sensitivity in peripheral tissues [92]. However, despite clear benefits in glycemic control, this class of drugs has recently fallen into disuse due to concerns over side effects and adverse events as extensively discussed by Soccio and colleagues in a recent review [93]. Results of available epidemiological studies on the incidence of cancer in rosiglitazonetreated patients are not univocal [94]. Evidence of both pro-carcinogenic and anti-tumorigenic effects of PPARγ agonists have been mainly reported in vitro and in vivo models that do not take into full account the metabolic characteristics of patients with diabetes. A population-based report published by Govindarajan and colleagues [95] showed that TZDs associates with reduced risk of lung cancer in diabetic patients in agreement with previous in vitro studies [96]. Ramos-Nino and colleagues showed an association between the use of TZDs and cancer in a community-based population of adults with diabetes participating in the Vermont Diabetes Information System (VDIS) [97]. The association was observed primarily among rosiglitazone users and not among subjects using pioglitazone. Furthermore, the association was found in women, but not in men. A case-control study published in 2013 showed that long-term exposure to rosiglitazone, or pioglitazone, increases the risk of bladder cancer in T2D patients [98].
Conclusion
Multiple hormones, growth factors, cytokines and other mediators associated with the obese state and the metabolic syndrome enable crosstalk between macrophages, adipocytes, endothelial cells and epithelial cells and contribute to cancer-related processes (including growth signaling, inflammation and vascular alterations) (Figure 1). Components of these interrelated pathways represent promising mechanism-based targets for the resolution of diabetes- induced cancer. Moreover, compounds that are beneficial for diabetes and cancer such as metformin represent a promising intervention that should be tested in future translational studies evaluating new strategies for breaking the diabetes-cancer link.
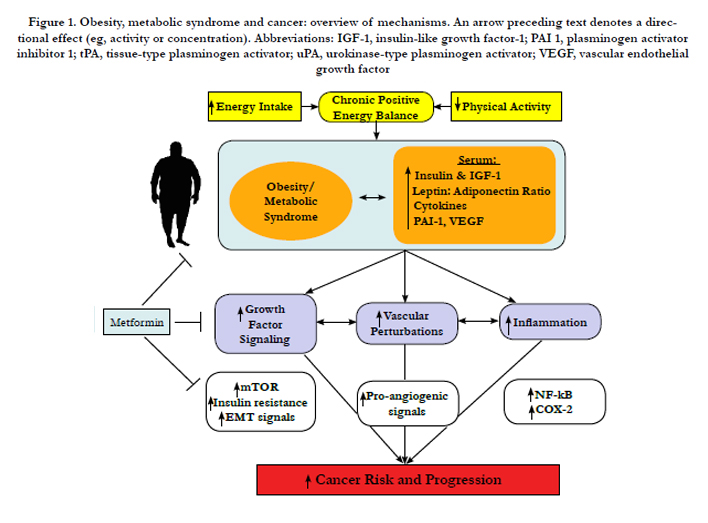
Figure 1. Obesity, metabolic syndrome and cancer: overview of mechanisms. An arrow preceding text denotes a directional effect (eg, activity or concentration). Abbreviations: IGF-1, insulin-like growth factor-1; PAI 1, plasminogen activator inhibitor 1; tPA, tissue-type plasminogen activator; uPA, urokinase-type plasminogen activator; VEGF, vascular endothelial growth factor
Acknowledgements
Dr. S. Hursting is funded, in part, by grants from the National Cancer Institute (R01CA129409), the Breast Cancer Research Foundation (UTA09-001068), and the National Institute of Environmental Health Sciences (P30ES007784). The authors have nothing to disclose.
References
- Flegal KM, Carroll MD, Ogden CL, Curtin LR (2010) Prevalence and trends in obesity among US adults, 1999-2008. JAMA 303(3): 235-241.
- Ervin RB (2009) Prevalence of metabolic syndrome among adults 20 years of age and over, by sex, age, race, and ethnicity, and body mass index: United States, 2003-2006. Natl Health Stat Report (13): 1-7.
- Hursting SD, Berger NA (2010) Energy balance, host-related factors, and cancer progression. J Clin Oncol 28(26): 4058-4065.
- Carter JC, Church FC (2009) Obesity and breast cancer: the roles of peroxisome proliferator-activated receptor-gamma and plasminogen activator inhibitor-1. PPAR Res 2009: 345320.
- Poirier P, Giles TD, Bray GA, Hong Y, Stern JS, et al (2006) Obesity and cardiovascular disease: pathophysiology, evaluation, and effect of weight loss. Arterioscler Thromb Vasc Biol 26(5): 968-976.
- World Cancer Research Fund/American Institute for Cancer Research (2007) Food, nutrition, physical activity and the prevention of cancer: a global perspective. Washington, DC.
- Colditz GA, Wolin KY, Gehlert S (2012) Applying what we know to accelerate cancer prevention. Sci Transl Med 4(127): 127rv4.
- Bower JE, Bak K, Berger A, Breitbart W, Escalante CP, et al. (2014) Screening, Assesment, and Management of Fatigue in Adult survivors of Cancer: An American Society of Clinical Oncology Clinical Practice Guideline Adaptation. J Clin Oncol 32(17): 1840-1850.
- Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ (2003) Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N Engl J Med 348(17): 1625-1638.
- CDC, (2011) National Diabetes Fact Sheet, Atlanta, GA. www.cdc.gov/diabetes/pubs/pdf/ndfs_2011.pdf
- Gallagher EJ, LeRoith D (2011) Diabetes, cancer, and metformin: connections of metabolism and cell proliferation. Ann N Y Acad Sci 1243:54-68.
- Everhart J, Wright D (1995) Diabetes mellitus as a risk factor for pancreatic cancer. A meta-analysis. JAMA 273(20): 1605-1609.
- Huxley R, Ansary-Moghaddam A, Berrington de Gonzalez A, Barzi F, Woodward M, et al. (2005) Type-II diabetes and pancreatic cancer: a metaanalysis of 36 studies. Br J Cancer 92(11): 2076-2083.
- Friberg E, Orsini N, Mantzoros CS, Wolk A (2007) Diabetes mellitus and risk of endometrial cancer: a meta-analysis. Diabetologia 50(7): 1365-1374.
- El-Serag HB, Hampel H, Javadi F (2006) The association between diabetes and hepatocellular carcinoma: a systematic review of epidemiologic evidence. Clin Gastroenterol Hepatol 4(3): 369-380.
- Larsson SC, Mantzoros CS, Wolk A (2007) Diabetes mellitus and risk of breast cancer: a meta-analysis. Int J Cancer 121(4): 856-862.
- Larsson SC, Orsini N, Brismar K, Wolk A (2006) Diabetes mellitus and risk of bladder cancer: a meta-analysis. Diabetologia 49(12): 2819-2823.
- MacKenzie T, Zens MS, Ferrara A, Schned A, Karagas MR (2011) Diabetes and risk of bladder cancer: evidence from a case-control study in New England. Cancer 117(7): 1552-1556.
- Larsson SC, Wolk A (2011) Diabetes mellitus and incidence of kidney cancer: a meta-analysis of cohort studies. Diabetologia 54(5): 1013-1018.
- Larsson SC, Orsini N, Wolk A (2005) Diabetes mellitus and risk of colorectal cancer: a meta-analysis. J Natl Cancer Inst 97(22): 1679-1687.
- Kasper JS, Giovannucci E (2006) A meta-analysis of diabetes mellitus and the risk of prostate cancer. Cancer Epidemiol Biomarkers Prev 15(11): 2056- 2062.
- Bonovas S, Filioussi K, Tsantes A (2004) Diabetes mellitus and risk of prostate cancer: a meta-analysis. Diabetologia 47(6): 1071-1078.
- Braun S, Bitton-Worms K, LeRoith D (2011) The link between the metabolic syndrome and cancer. Int J Biol Sci 7(7): 1003-1015.
- Pollak M (2008) Insulin and insulin-like growth factor signalling in neoplasia. Nat Rev Cancer 8(12): 915-928.
- Hursting SD, Smith SM, Lashinger LM, Harvey AE, Perkins SN (2010) Calories and carcinogenesis: lessons learned from 30 years of calorie restriction research. Carcinogenesis 31(1): 83-89.
- Chao W, D’Amore PA (2008) IGF2: epigenetic regulation and role in development and disease. Cytokine Growth Factor Rev 19(2): 111-120.
- Pandini G, Frasca F, Mineo R, Sciacca L, Vigneri R, et al. (2002) Insulin/ insulin-like growth factor I hybrid receptors have different biological characteristics depending on the insulin receptor isoform involved. J Biol Chem 277(42): 39684-39695.
- Mosthaf L, Vogt B, Häring HU, Ullrich A (1991) Altered expression of insulin receptor types A and B in the skeletal muscle of non-insulin-dependent diabetes mellitus patients. Proc Natl Acad Sci USA 88(11): 4728-4730.
- Sesti G, Marini MA, Tullio AN, Montemurro A, Borboni P, et al. (1991) Altered expression of the two naturally occurring human insulin receptor variants in isolated adipocytes of non-insulin-dependent diabetes mellitus patients. Biochem Biophys Res Commun 181(3): 1419-1424.
- Janssen JA, Varewijck AJ (2014) IGF-IR Targeted Therapy: Past, Present and Future. Front Endocrinol (Lausanne) 5: 224.
- Wong KK, Engelman JA, Cantley LC (2010) Targeting the PI3K signaling pathway in cancer. Curr Opin Genet Dev 20(1): 87-90.
- Memmott RM, Dennis PA (2009) Akt-dependent and -independent mechanisms of mTOR regulation in cancer. Cell Signal 21: 656-664.
- Lindsley JE, Rutter J (2004) Nutrient sensing and metabolic decisions. Comp Biochem Physiol B Biochem Mol Biol 139(4): 543-559.
- Avruch J, Hara K, Lin Y, Liu M, Long X, et al. (2006) Insulin and aminoacid regulation of mTOR signaling and kinase activity through the Rheb GTPase. Oncogene 25(48): 6361-6372.
- Gautron L, Elmquist JK (2011) Sixteen years and counting: an update on leptin in energy balance. J Clin Invest 121: 2087-2093.
- Villanueva EC, Myers MG (2008) Leptin receptor signaling and the regulation of mammalian physiology. Int J Obes 32(Suppl 7): S8-S12.
- Vaiopoulos AG, Marinou K, Christodoulides C, Koutsilieris M (2012) The role of adiponectin in human vascular physiology. Int J Cardiol 155(2): 188-193.
- Barb D, Williams CJ, Neuwirth AK, Mantzoros CS (2007) Adiponectin in relation to malignancies: a review of existing basic research and clinical evidence. Am J Clin Nutr 86(3): S858-S866.
- Stofkova A (2009) Leptin and adiponectin: from energy and metabolic dysbalance to inflammation and autoimmunity. Endocr Regul 43(4): 157-168.
- Stattin P, Lukanova A, Biessy C, Soderberg S, Palmqvist R, et al. (2004) Obesity and colon cancer: does leptin provide a link? Int J Cancer 109: 149-152.
- Chang S, Hursting SD, Contois JH, Strom SS, Yamamura Y, et al. (2001) Leptin and prostate cancer. Prostate 46(1): 62-67.
- Wu MH, Chou YC, Chou WY, Hsu GC, Chu CH, et al. (2009) Circulating levels of leptin, adiposity and breast cancer risk. Br J Cancer 100(4):578-582.
- Fenton JI, Hord NG, Lavigne JA, Perkins SN, Hursting SD (2005) Leptin, insulin-like growth factor-1, and insulin-like growth factor-2 are mitogens in ApcMin/+ but not Apc+/+ colonic epithelial cells. Cancer Epidemiol Biomarkers Prev 14(7): 1646-1652.
- Grossmann ME, Nkhata KJ, Mizuno NK, Ray A, Cleary MP (2008) Effects of adiponectin on breast cancer cell growth and signaling. Br J Cancer 98(2):370-379.
- Rzepka-Gorska I, Bedner R, Cymbaluk-Ploska A, Chudecka-Glaz A (2008) Serum adiponectin in relation to endometrial cancer and endometrial hyperplasia with atypia in obese women. Eur J Gynaecol Oncol 29(6): 594-597.
- Harvey AE, Lashinger LM, Hursting SD (2011) The growing challenge of obesity and cancer: an inflammatory issue. Ann NY Acad Sci 1229(1): 45-52.
- Olefsky JM, Glass CK (2010) Macrophages, inflammation, and insulin resistance. Annu Rev Physiol 72: 219-246.
- O'Rourke RW (2009) Inflammation in obesity-related diseases. Surgery 145(3): 255-259.
- Renehan AG, Roberts DL, Dive C (2008) Obesity and cancer: pathophysiological and biological mechanisms. Arch Physiol Biochem 114(1): 71-83.
- Karin M (2006) Nuclear factor-kappaB in cancer development and progression. Nature 441: 431-436.
- Balkwill F, Mantovani A (2001) Inflammation and cancer: back to Virchow? Lancet 357(9255): 539-545.
- Foltz CJ, Fox JG, Cahill R, Murphy JC, Yan L, et al. (1998) Spontaneous inflammatory bowel disease in multiple mutant mouse lines: association with colonization by Helicobacter hepaticus. Helicobacter 3(2): 69-78.
- Coussens LM, Werb Z (2002) Inflammation and cancer. Nature 420(6917): 860-867.
- Allavena P, Sica A, Garlanda C, Mantovani A (2008) The Yin-Yang of tumor- associated macrophages in neoplastic progression and immune surveillance.Immunol Rev 222(1): 155-161.
- Koki A, Khan NK, Woerner BM, Dannenberg AJ, Olson L, et al. (2002) Cyclooxygenase-2 in human pathological disease. Adv Exp Med Biol 507: 177-184.
- Iwaki T, Urano T, Umemura K (2012) PAI-1, progress in understanding the clinical problem and its aetiology. Br J Haematol 157(3): 291-298.
- Byrne AM, Bouchier-Hayes DJ, Harmey JH (2005) Angiogenic and cell survival functions of vascular endothelial growth factor (VEGF). J Cell Mol Med 9(4): 777-794.
- Liu Y, Tamimi RM, Collins LC, Schnitt SJ, Gilmore HL, et al. (2011) The association between vascular endothelial growth factor expression in invasive breast cancer and survival varies with intrinsic subtypes and use of adjuvant systemic therapy: results from the Nurses’ Health Study. Breast Cancer Res Treat 129(1): 175-184.
- Cao Y (2007) Angiogeneis modulates adipogenesis and obesity. J Clin Invest 117(9): 2362-2368.
- Renehan AG (2010) Body fatness and bevacizumab-based therapy in metastatis colorectal cancer. Gut 59(3): 289-290.
- Simkens LH, Koopman M, Mol L, Veldhuis GJ, Ten Bokkel Huinink D, et al. (2011) Influence of body mass index on outcome in advanced colorectal cancer patients receiving chemotherapy with or without targeted therapy. Eur J Cancer 47(17): 2560-2567.
- Giovannucci E, Harlan DM, Archer MC, Bergenstal RM, Gapstur SM, et al. (2010) Diabetes and cancer: a consensus report. Diabetes Care 33: 1674- 1685.
- Joslin EP, Lombard HL, Burrows RE, Manning MD (1959) Diabetes and Cancer. N Engl J Med 260(10): 486-488.
- Bao B, Wang Z, Li Y, Kong D, Ali S, et al. (2011) The complexities of obesity and diabetes with the development and progression of pancreatic cancer. Biochim Biophys Acta 1815(2): 135-146.
- Becker S, Dossus L, Kaaks R (2009) Obesity related hyperinsulinaemia and hyperglycaemia and cancer development. Arch Physiol Biochem 115(2): 86-96.
- Bruce WR, Wolever TM, Giacca A (2000) Mechanisms linking diet and colorectal cancer: the possible role of insulin resistance. Nutr Cancer 37(1): 19-26.
- Giovannucci E, Michaud D (2007) The role of obesity and related metabolic disturbances in cancers of the colon, prostate, and pancreas. Gastroenterology 132(6): 2208-2225.
- Busik JV, Hootman SR, Greenidge CA, Henry DN (1997) Glucose-specific regulation of aldose reductase in capan-1 human pancreatic duct cells in vitro. J Clin Invest 100(7): 1685-1692.
- Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T, et al. (2000) Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 404(6779): 787-790.
- Cullen JJ, Mitros FA, Oberley LW (2003) Expression of antioxidant enzymes in diseases of the human pancreas: another link between chronic pancreatitis and pancreatic cancer. Pancreas 26(1): 23-27.
- Nathan DM, Buse JB, Davidson MB, Ferrannini E, Holman RR, et al. (2009) Medical management of hyperglycemia in type 2 diabetes: a consensus algorithm for the initiation and adjustment of therapy: a consensus statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care 32(1): 193-203.
- Currie CJ, Poole CD, Gale EA (2009) The influence of glucose-lowering therapies on cancer risk in type 2 diabetes. Diabetologia 52(9): 1766-1777.
- Foretz M, Hebrard S, Leclerc J, Zarrinpashneh E, Soty M, et al. (2010) Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J Clin Invest 120: 2355-2369.
- Kahn BB, Alquier T, Carling D, Hardie DG (2005) AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab 1(1): 15-25.
- Zhou G, Myers R, Li Y, Chen Y, Shen X, et al. (2001) Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest 108(8): 1167-1174.
- Marshall S (2006) Role of insulin, adipocyte hormones, and nutrient-sensing pathways in regulating fuel metabolism and energy homeostasis: a nutritional perspective of diabetes, obesity, and cancer. Sci STKE 2006(346): re7.
- Algire C, Zakikhani M, Blouin MJ, Shuai JH, Pollak M (2008) Metformin attenuates the stimulatory effect of a high-energy diet on in vivo LLC1 carcinoma growth. Endocr Relat Cancer 15(3): 833-839.
- Zakikhani M, Dowling R, Fantus IG, Sonenberg N, Pollak M (2006) Metformin is an AMP kinase-dependent growth inhibitor for breast cancer cells. Cancer Res 66: 10269-10273.
- Alimova IN, Liu B, Fan Z, Edgerton SM, Dillon T,et al. (2009) Metformin inhibits breast cancer cell growth, colony formation and induces cell cycle arrest in vitro. Cell Cycle 8: 909-915.
- Liu B, Fan Z, Edgerton SM, Deng XS, Alimova IN, at al. (2009) Metformin induces unique biological and molecular responses in triple negative breast cancer cells. Cell Cycle 8(13): 2031-2040.
- Bartel DP (2004) MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116(2): 281-297.
- Gregory PA, Bracken CP, Bert AG, Goodall GJ (2008) MicroRNAs as regulators of epithelial-mesenchymal transition. Cell cycle 7(20): 3112-3118.
- Cifarelli V, Lashinger LM, Devlin KL, Dunlap SM, Huang J (2015) Metformin and Rapamycin Reduce Pancreatic Cancer Growth in Obese Prediabetic Mice by Distinct MicroRNA-Regulated Mechanisms. Diabetes 64(5):1632-1642.
- Kashat M, Azzouz L, Sarkar SH, Kong D, Li Y, et al. (2012) Inactivation of AR and Notch-1 signaling by miR-34a attenuates prostate cancer aggressiveness. Am J Transl Res 4(4): 432-442.
- Ji Q, Hao X, Zhang M, Tang W, Yang M, et al. (2009) MicroRNA miR- 34 inhibits human pancreatic cancer tumor-initiating cells. PloS One 4(8): e6816.
- Jamieson NB, Morran DC, Morton JP, Ali A, Dickson EJ, et al. (2012) MicroRNA molecular profiles associated with diagnosis, clinicopathologic criteria, and overall survival in patients with resectable pancreatic ductal adenocarcinoma. Clin Cancer Res 18(2): 534-545.
- Zhang J, Tian XJ, Zhang H, Teng Y, Li R, et al. (2014) TGF-beta-induced epithelial-to-mesenchymal transition proceeds through stepwise activation of multiple feedback loops. Sci Signal 7(345): ra91.
- Niessen K, Fu Y, Chang L, Hoodless PA, McFadden D, et al. (2008) Slug is a direct Notch target required for initiation of cardiac cushion cellularization. J Cell Biol 182(2): 315-325.
- Wang Z, Li Y, Banerjee S, Sarkar FH (2009) Emerging role of Notch in stem cells and cancer. Cancer Lett 279(1): 8-12.
- Leong KG, Niessen K, Kulic I, Raouf A, Eaves C, et al. (2007) Jagged1- mediated Notch activation induces epithelial-to-mesenchymal transition through Slug-induced repression of E-cadherin. J Exp Med 204(12): 2935-2948.
- Sahlgren C, Gustafsson MV, Jin S, Poellinger L, Lendahl U (2008) Notch signaling mediates hypoxia-induced tumor cell migration and invasion. Proc Natl Acad Sci USA 105(17): 6392-6397.
- Vamecq J, Latruffe N (1999) Medical significance of peroxisome proliferator- activated receptors. Lancet 354(9173): 141-148.
- Soccio RE, Chen ER, Lazar MA (2014) Thiazolidinediones and the promise of insulin sensitization in type 2 diabetes. Cell Metab 20(4): 573-591.
- Monami M, Lamanna C, Marchionni N, Mannucci E (2008) Rosiglitazone and risk of cancer: a meta-analysis of randomized clinical trials. Diabetes Care 31(7): 1455-1460.
- Govindarajan R, Ratnasinghe L, Simmons DL, Siegel ER, Midathada MV, et al. (2007) Thiazolidinediones and the risk of lung, prostate, and colon cancer in patients with diabetes. J Clin Oncol 25(12): 1476-1481.
- Han S, Roman J (2006) Rosiglitazone suppresses human lung carcinoma cell growth through PPARgamma-dependent and PPARgamma-independent signal pathways. Mol Cancer Ther 5(2): 430-437.
- Ramos-Nino ME, MacLean CD, Littenberg B (2007) Association between cancer prevalence and use of thiazolidinediones: results from the Vermont Diabetes Information System. BMC Med 5: 17.
- Hsiao FY, Hsieh PH, Huang WF, Tsai YW, Gau CS (2013) Risk of bladder cancer in diabetic patients treated with rosiglitazone or pioglitazone: a nested case-control study. Drug Safe 36(8): 643-649.





