Genetic Determinants of Type 2 Diabetes in Asians
Qi Q1*, Wang X1,2, Strizich G1, Wang T1
1. Department of Epidemiology and Population Health, Albert Einstein College of Medicine, Bronx, NY 10461, USA.
2. Department of Epidemiology and Biostatistics, School of Public Health, Peking University Health Science Center, 38 Xueyuan Road, Haidian District, Beijing 100191, China
*Corresponding Author
Dr. Qi Q,
Department of Epidemiology and Population Health,
Albert Einstein College of Medicine,
1300 Morris Park Avenue, Belfer 1306A Bronx,
NY 10461, USA.
Tel: 718-430-4203
Fax: 718-430-8653
E-mail: qibin.qi@einstein.yu.edu
Article Type: Review Article
Recieved: December 15, 2014; Accepted: March 10, 2015; Published: March 12, 2015
Citation: Qi Q, Wang X, Strizich G, Wang T (2015) Genetic Determinants of Type 2 Diabetes in Asians. Int J Diabetol Vasc Dis Res, S1:001 1-9.doi: dx.doi.org/10.19070/2328-353X-SI01001
Copyright: Qi Q© 2015. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
Abstract
Type 2 diabetes (T2D) has become a major health problem throughout the world and the epidemic is particularly severe in Asian countries. Compared with European populations, Asians tend to develop diabetes at a younger ageand at much higher incidence rates given the same amount of weight gain. Genome-wide association studies (GWAS) have identified over 70 loci associated with T2D. Although the majority of GWAS results were conducted in populations of European ancestry, recent GWAS in Asians have made important contributions to the identification of T2D susceptibility loci. These studies not only confirmed T2D susceptibility loci initially identified in European populations, but also identified novel susceptibility loci that provide new insights into the pathophysiology of diseases. In this article, we review GWAS results of T2D conducted in East and South Asians and compare them to thoseof European populations. Currently identified T2D genetic variants do not appear to explain the phenomenon that Asians are more susceptible to T2D than European populations, suggesting further studies in Asian populations are needed.
2.Genetic studies of T2D prior to GWAS
3.GWAS for T2D
3.1 East Asians
3.2 South Asians
4.Comparison of T2D susceptibility loci between European and Asian populations
5.Copy Number Variants
6.Thrifty Genotype
7.Conclusion and Future Perspective
8.Acknowledgements
9.References
Introduction
Type 2 diabetes (T2D) has become a leading health problem throughout the world and the epidemic is particularly notable in developing countries. According to estimates by the International Diabetes Federation, the total number of people with diabetes worldwide is projected to rise from 366 million to 552 million by the year 2030, with two-thirds of all new diabetes cases occurring in low- to middle-income countries [1]. Accounting for roughly 60% of the world’s population, Asia’s rapid economic development and urbanization have made it an epicenter of the epidemic [2], with explosive increases in diabetes prevalence in recent decades [3]. In 1980, for example, less than 1% of Chinese adults had T2D. By 2008, the prevalence had reached nearly 10%, or more than 92 million Chinese adults, and another 148 million were prediabetic [4]. Compared with European populations, Asians develop diabetes at younger ages, and at much higher rates given the same amount of weight gain [2]. Several factors contribute to the accelerated diabetes epidemic in Asians, including a high prevalence of smoking and heavy alcohol use; high intake of refined carbohydrates (e.g. white rice); and dramatically lower physical activity levels [2].
It has long been recognized that there are strong genetic influences of T2D, as revealed through classical genetic research, including twin, adoption, and family studies. With the rapid development of modern genotyping techniques, a number of T2D loci have been identified and established by genome-wide association studies (GWAS) among the world’s major ethnic populations, mostly in European and Asian populations. In this article, we aimed to summarize recent progress on the GWAS of T2D in Asians. We also compared these identified T2D susceptibility loci between European and Asian populationsanddiscussed whether currently known genetic variants can explain ethnic differences in T2D risk.
Genetic studies of T2D prior to GWAS
Candidate gene and genome-wide linkage studies (or large-scale association studies), the two major approaches for identifying genes that predispose to common complex diseases before the GWAS era, were limited by small sample sizes and lack of replication of results [5,6]. In Asian populations, despite identification of numerous T2D susceptibility loci through candidate gene approach, few went on to be validated in other studies. The first signals associated with T2D to be robustly replicated in European populations were the P12 A polymorphism (rs1801282) in PPARG and E23K (rs5219) polymorphism in KCNJ11 [7, 8]. Recent meta-analyses have confirmed the associations of these genetic variants with risk of T2D in Asian populations [9-11]. TCF7L2, the first T2D susceptibility gene identified by largescale association analysis in European populations [12], has also been confirmed as a sucseptibility locus in Asian populations [16, 17]. However, the strong association between common variant rs 7903146 in TCF7L and risk of T2D that was highly confirmed in numerous European replication studies and GWAS [13-15], has not been replicated in Asians. Several studies in Han Chinese have reported different genetic variants in this locus associated with T2D [18, 19].
GWAS for T2D
With rapid improvements in high-throughput single nucleotide polymorphisms (SNPs) genotyping technology and development of the Hap Map project, methods for identifying susceptibility genes have changed dramatically. The GWAS is currently the most commonly-used approach for uncovering novel loci associated with T2D and related traits. To date, over 70 loci have been associated with T2D at a genome-wide significance level (P<5×10-8). Although the majority of existing GWAS of T2D have been conducted among populations of European ancestry [13-15, 20-32], more recent GWAS in Asians have also successfully identified a number of novel T2D loci [33-45] (Table 1).
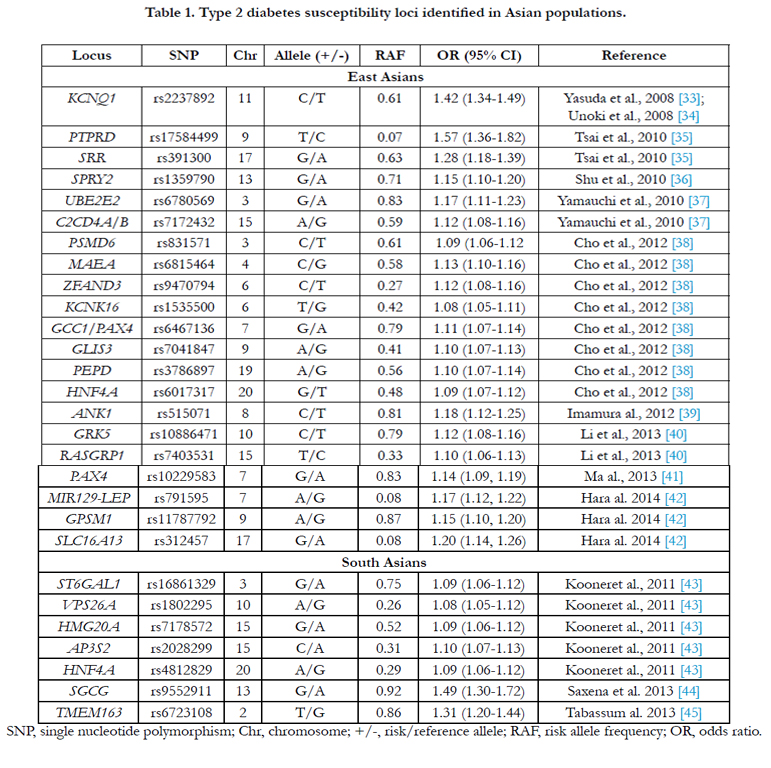

Table 1. Type 2 diabetes susceptibility loci identified in Asian populations.
SNP, single nucleotide polymorphism; Chr, chromosome; +/-, risk/reference allele; RAF, risk allele frequency; OR, odds ratio.
In 2008, two independent groups from Japan concurrently reported the first GWAS for T2D in Asians. They identified KCNQ1 as a new T2D susceptibility locus in East Asians, with an odds ratio (OR) of 1.42 per risk allele [33, 34]. The association was subsequently confirmed in European-descent populations [33, 34] KCNQ1 encodes the pore-forming subunit of a voltage-gated K+ channel that plays a key role for the repolarization of the cardiac action potential as well as water and salt transport in epithelial tissues [46]. It is also expressed in pancreatic islet cells and has been suggested to be involved in the regulation of insulin secretion [47]. Several subsequent studies confirming the association between KCNQ1 variants and risk of T2D in East Asian populations have suggested that KCNQ1 variants might confer T2D risk through impaired beta-cell function [48-50]. A second, independent signal in this locus was identified in European populations by ameta-analysis in the Diabetes, Genetics, Replication and Metaanalysis Consortium (DIAGRAM) [31].
In 2010, Tsai et al. [35] reported a two-stage GWAS for T2D in Han Chinese from Taiwan. They confirmed the previously reported association between KCNQ1 and T2D, and also identified two novel T2D susceptibility loci, PTPRD and SRR. PTPRD belongs to the receptor type IIA (R2A) subfamily of protein tyrosine phosphatases (PTPs), which have been implicated in neural development, cancer, and diabetes [51]. SRR encodes a serine racemase that synthesizes D-serine from L-serine and may play a role in regulating insulin and/or glucagon secretion through glutamate signaling in the pancreas [52]. In addition, Shu et al. [36] reported another novel T2D susceptibility locus, SPRY2, in a multistage- GWAS in Chinese populations. They also found an independent genetic variant (rs10906115) near CDC123, a known T2D locus previously identified in European populations [15] at the genomewide significance level. SPRY2 encodes a protein belonging to the sprouty family and inhibits receptor tyrosine kinase-induced signaling and is required for growth factor stimulated translocation of the protein to membrane ruffles. SPRY4, a homolog of SPRY2, inhibits the insulin receptor-transduced MAPK signaling pathway and regulates development of the pancreas [53].
Yamauchi et al. [37] conducted a three-stage GWAS for T2D in a Japanese population and identified T2D susceptibility loci at UBE2E2 and C2CD4A/B. UBE2E2 encodes the ubiquitinconjugating enzyme E2E2, and it has been suggested that the ubiquitin-proteasome system may play important roles in insulin secretion [54]. Indeed, the risk allele of UBE2E2 SNP rs7612463 was associated with lower homeostasis model assessment of betacell function (HOMA-β) among 872 non-diabetic control subjects [37]. C2CD4A/B encodes C2 calcium-dependent domain containing proteins 4A and 4B, nuclear factors with a role in regulating genes that control cellular architecture [55]. However, evidence for a role of C2CD4A/B in conferring susceptibility to T2D is lacking. Of note, follow-up studies in European populationsconfirmed the C2CD4A/B locus to be associated with T2D, whereas the UBE2E2 locus was not associated with T2D [37].
In 2012, Cho et al. [38] conducted a meta-analysis of eight T2D GWAS followed by 2-stage replication analyses with a total of roughly 25,000 cases and 30,000 controls in East Asian populations.The combined analysis identified eight new T2D loci reaching genome-wide significance level, mapping in or near GLIS3, PEPD, FITM2-R3HDML-HNF4A, KCNK16, MAEA, GCC1- PAX4, PSMD6, and ZFAND3.PEPD, encoding peptidase D involved in insulin secretion [56], as well as, HNF4A, encoding hepatocyte nuclear factor 4 alpha, have been reported to be associated with maturity-onset diabetes of the young type 1 (MODY1) diseases and T2D [57-60]. GLIS3 encodes a Krüppel-like zinc finger transcription factor that has been proposed as a key player in the regulation of pancreatic beta cell development and insulin gene expression [61]. KCNK16 encodes a potassium channel protein containing two pore-forming P domains, and potassium channels inhibited by ATP regulate glucose-dependent insulin secretion in pancreatic beta-cells [62]. MAEA encodes a protein with a role in erythroblast enucleation and in the development of mature macrophages [63]. GCC1 encodes a GRIP-domain–containing protein that might have a role in the organization of the trans-Golgi network [64]. PAX4 encodes paired box 4 which is involved in pancreatic islet development [65]. PSMD6 encodes 26S proteasome non-ATPase regulatory subunit 6, which is probably involved in the ATP-dependent degradation of ubiquitinated proteins [66]. The function of ZFAND3, which encodes zinc finger AN1-type domain 3, has not been fully elucidated.
Imamura et al. [39] conducted an imputation-based GWAS in Japanese populations and reported ANK1 as a new T2D susceptibility locus. ANK1 encodes a member of the ankyrin family which plays a critical role in stabilizing the membrane structure of erythrocytes [67]. The role of the gene in T2D was further validated in a recent study demonstrating that two additional SNPs in ANK1 were associated with HbA1c levels in European, nondiabetic adults [68].
In 2013, Li et al. [40] performed a three-stage GWAS for T2D in a total of 8,569 cases and 8,923 controls of Han Chinese, followed by an in silico replication in the aforementioned East Asian meta-analysis [38]. This analysis confirmed seven T2D loci previously identified in Europeans (such as CDKAL1, CDKN2A/B, CDC123, HNF1B, and DUSP9) and East Asians (such as KCNQ1 and GLIS3) at genome-wide significance, and identified two novel T2D loci at GRK5 and RASGRP1. The T2D risk allele of RASGRP1 was also associated with higher HbA1c and lower homeostasis model assessment of beta-cell function, whereas the T2D risk allele of GRK5 was associated with higher fasting insulin. GRK5 encodes G-protein coupled receptor (GPCR) kinase 5 which plays a crucial role in phosphorylation of multiple GPCRs and non-GPCR substrates. These receptors and substrates, such as glucagon receptor [69], b2-adrenergic receptor [70, 71], Hsp70-interacting protein [72], and nuclear factor-kB1/p105 [73], are all important regulators of glucose homeostasis orinflammation. RASGRP1 encodes the RAS guanyl releasing protein1 which is involved in the development and function of lymphocytes. Its dysfunction in beta-cells was also reported to lead to islet inflammation and impaired beta-cell function [74].
A further GWAS conducted in Southern Han Chinese by Ma et al. [41], identified a novel T2D locus at 7q32 near PAX4, with successful replications in other Chinese and East Asians. The risk allele of the PAX4 variant was also associated with elevated fasting glucose and impaired beta-cell function. PAX4 plays an important role in the differentiation and development of pancreatic beta cells [75]. Previous studies have reported that diabetic patients carrying PAX4 mutation have serious defects in first-phase insulin secretion [76], and mutations in PAX4 may cause rare monogenic forms of young-onset diabetes, including maturity-onset diabetes of the young type 9 (MODY9)diseases [77].
In 2014, Hara et al. [42] performed a three-stage GWAS in Japanese and other East Asians where they identified three new loci for T2D: MIR129/LEP, GPSM1 and SLC16A13. Leptin, encoded by LEP, is responsible for regulating of body weight by inhibiting food intake and stimulating energy expenditure [78]. GPSM1 might be a biologically plausible obesity gene with the evidence that Gpsm1 null mice have a lean phenotype with reduced fat mass and increased nocturnal energy expenditure [79]. Intestinal expression of SLC16A13 has been suggested to be upregulated by peroxisome proliferator-activated receptor-αagonists [80]. Interestingly, there was no association between these three novelloci and T2D in European populations [81].
In 2011, Kooner et al. [43] reported the first GWAS for T2D in cases and controls with ancestry from the Indian Subcontinent (India, Pakistan, Sri Lanka, and Bangladesh). They identified six T2D susceptibility loci: GRB14, ST6GAL1, VPS26A, HMG20A, AP3S2, and HNF4A. They also reported thatSNPs at GRB14 were associated with insulin sensitivity, andSNPs at ST6GAL1 and HNF4A with beta-cell function [43]. GRB14 encodes growth factor receptor-bound protein 14, an adaptor protein that binds to insulin receptors and insulin-like growth-factor receptors to inhibit tyrosine kinase signaling [82]. ST6GAL1, encoding an enzyme predominantly located in the Golgi apparatus, is involved in post-translational glycosylation of cell-surface components. Glycosylation through addition of sialic acid residues is reported to influence both insulin action and cell surface trafficking [83].
In 2013, Saxena et al. [44] conducted a GWAS and multistage meta- analysis in Punjabi Sikhs from Northern India. They identified a novel locus at 13q12 in the SGCG gene associated with T2D. Interestingly, the associated SNP is monomorphic in Europeans, suggesting this locus might be specific to the Indian Punjabi Sikh population. SGCG is a member of the sarcoglycan complex of transmembrane glycoproteins mutated in autosomal recessive muscular dystrophy [44]. Mice lacking sarcoglycan complex in adipose and skeletal muscle have been shown to be glucose intolerant and displayed whole body insulin resistance because of impaired insulin-stimulated glucose uptake in skeletal muscle [84]. In addition, Tabassum et al. [45], through a two-stage GWAS in Indians, reported a new T2D-associated locus in TMEM163 gene at 2q21. TMEM163, encoding a synaptic vesicle membrane protein with six putative transmembrane helices, has been shown to play an important role in reducing fasting insulin levels and HOMA-IR [45].
Comparison of T2D susceptibility loci between European and Asian populations
Most T2D loci initially identified in European populations have been replicated in Asians [38, 43]. Kooner et al. [43] investigated 42 T2D loci in both South Asians and Europeans, and found that 37 loci showed consistent direction of effect in South Asians and Europeans, and that 27 loci were associated with T2D at P< 0.05 in South Asians. Top signals observed in the East Asian T2D stage 1 meta-analysis [38], such as CDKAL1, CDKN2A/B, HHEX/ IDE, KCNJ11, KCNQ1, and FTO, were similar to those identified in European populations (the DIAGRAM+ stage 1 metaanalysis) [31], though additional genetic loci have been identified in European populations that have not been tested in Asians. In addition, a GWAS of T2D in multi-ethnic cohorts from Southeast Asia (including Chinese, Malays and Asian Indians) observedconsistent direction of effect for many T2D SNPs identified in European populations [85].
However, there were some inter-ethnic differences in the risk allele frequencies (RAFs) and genetic variant locations of top T2D loci among European and South and East Asian populations. With the exception of SNPs in CDKAL1 and KCNQ1, the RAFs of most SNPs in East Asians are lower than those in Europeans and South Asians (Figure 1). For example, the risk allele of the SNP rs7903146 in TCF7L2 is common in European and South Asian populations (20-30%), but rare in East Asians (~2%). Other common genetic variants in TCF7L2 (e.g., SNPs rs290487 and rs11196218) with RAFs of 30-40% were reported to confer risk for T2D that is exclusive to the Chinese population [18, 19].
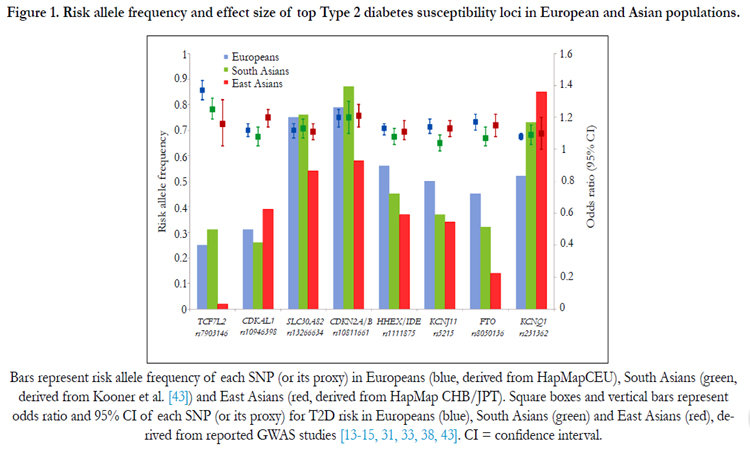
Figure 1. Risk allele frequency and effect size of top Type 2 diabetes susceptibility loci in European and Asian populations
Bars represent risk allele frequency of each SNP (or its proxy) in Europeans (blue, derived from HapMapCEU), South Asians (green, derived from Kooner et al. [43]) and East Asians (red, derived from HapMap CHB/JPT). Square boxes and vertical bars represent odds ratio and 95% CI of each SNP (or its proxy) for T2D risk in Europeans (blue), South Asians (green) and East Asians (red), derived from reported GWAS studies [13-15, 31, 33, 38, 43]. CI =confidence interval.
Despite heterogeneity in risk allele frequency of genetic variants with the strongest T2D association signals, the effect sizes appear to be similar across populations. Only a handful of SNPs showed potential heterogeneity between European and Asian populations (Figure 1). The association between TCF7L2SNP rs7903146 and risk of T2D is stronger in Europeans (odds ratio [OR] 1.37 [1.31- 1.43]) compared to East Asians (OR 1.16 [1.02-1.31]; P for heterogeneity =0.009 between Europeans and East Asians), but there is no significant difference between East and South Asians. In contrast, CDKAL1 variants appear to introduce a stronger effect in East Asians than in Europeans. A multi ethnic cohort study, for example, reported that the genetic effect of CDKAL1 on T2D was stronger in Japanese Americans than European Americans [86]. Other studies have observed similarly stronger associations among East Asians compared to Europeans [56, 87]. By comparing the data from three large-scale meta-analyses [31, 38, 43], the magnitude of the effect of CDKAL1 for T2D is stronger in East Asians (OR 1.20 [1.14-1.25]) compared to Europeans (OR 1.12 [1.08-1.16], P for heterogeneity = 0.02 between East Asians and Europeans) and South Asians (OR 1.08 [1.02-1.14], P for heterogeneity = 0.004 between East and South Asians).
Furthermore, several combined analyses of multiple genetic variants using genetic risk scores in Asians have shown similar results with those in European populations, in which each risk allele increment was associated with a 10-20% increased risk of T2D [88-95]. For example, Meigs et al. [88] investigated the combined effect of 18 T2D genetic variants in participants of European ancestry from the Framingham Off spring Study and found that the OR per risk allele for T2D was 1.12. Similarly, in a combined analysis of 17 T2D genetic variants in a population-based Han Chinese cohort, each risk allele was associated with an 18% increased risk for T2D [93]. More recently, Janipalli et al. [95] reported an OR per risk allele of 1.11 for T2D in Indians by using a genetic risk score based on 32 T2D genetic variants. These data suggest that the overall contribution of the identified genetic loci to T2D is similar between European and Asian populations and that these genetic loci do not appear to explain ethnic differences in diabetes risk.
Veryrecently, a trans-ethnic meta-analysis of GWAS evaluated 69 established T2D loci among four major ethnic groups (European, East Asian, South Asian and Mexican ancestry) and found significant excess in directional consistency of T2D risk alleles across ancestry groups, with only 3 loci showing nominal evidence of heterogeneity [96]. Specifically, the effect of TCF7L2 (rs7903146) is strongest in populations of European ancestry, whereas the association signals of PEPD (rs3786897) and KLF14 (rs3786897) are largely specific to East Asian and European populations, respectively. In addition, seven newly-identified T2D susceptibility loci without heterogeneity across ethnic groups were identified, including TMEM154, POU5F1/TCF19, ARL15,SSR1/RREB1, MPHOSPH9,FAF1 and LPP.
Copy Number Variants
In addition to SNPs, copy number variants (CNVs) also account for a major proportion of human genetic variation and may have an important role in genetic susceptibility to common diseases. However, to date there are few reported associations between common CNVs and obesity and T2D [97-99]. In 2010, the Well come Trust Case Control Consortium performed a GWAS between CNVs and eight common human diseases in European populations, confirming three loci where CNVs were associated with Crohn’s disease (IRGM locus), rheumatoid arthritis and type 1 diabetes (HLA loci), and T2D (TSPAN8 locus) [98]. Each locus had previously been identified in SNP-based studies. Another GWAS of CNVs, however, failed to observe robust associations between CNVs and the risk of T2D in a Korean population [99]. These analyses suggest that common CNVs on existing platforms are not likely to have major contributions to T2D genetic susceptibility. However, some functional CNVs associated with gene expression levels are still potential candidates. One example where copy number has been shown to play a role comes from the salivary amylase gene (AMY1), whose copy number has been positively correlated with salivary amylase protein level [100]. Individuals from populations with high-starch diets have more AMY1 copies than those with traditionally low-starch diets [100]. This suggests a positive selection of AMY1 copies through a dietary shift early during human evolutionary history, especially in some ethnic groups such as East Asians who consume high-starch diets. Although there is no evidence for an association between AMY1 copies and T2D, this example could provide new insights to guide future genetic association studies for T2D where by differences in diet or environment exposures and the evolution of human genes among different ethnic groups are considered.
Thrifty Genotype
In 1962, Neel [101] suggested that famine exposure during human evolutionary history resulted in positive selection for thrifty genotypes characterized by high efficiency of energy metabolism and fat storage during periods of feast. Thus, obesity and T2D might be a result of a thrifty genotype that leads to disadvantageous phenotypes in the modern setting of food over abundance and sedentary life styles. The thrifty genotype hypothesis has been widely used to explain the extraordinarily high rates of diabetes seen among some indigenous populations such as Pima Indians of North America, since these populations may have an enhanced genetic predisposition to obesity and diabetes due to evolutionary selection for thrifty genotypes. In contrast, Europeans are thought less likely to possess thrifty genotypes as they evolved in environments that were less affected by feast and famine cycles. However, based on an analysis of 30 SNP data (17 T2D loci and 13 obesity loci) among African, European and East Asian populations from Hap Map, Southam et al. [102] found no clear evidence for over representation of derived alleles (versus ancestral alleles) for the T2D or obesity loci,nor did they find a greater concentration of these loci in a particular ethnic group. Moreover, in a recent analysis of 30 SNPs in 16 T2D loci and 28 SNPs in obesity loci among 53 populations from the Human Genome Diversity Panel, Klimentidis et al. [103] found no evidence that risk alleles associated with either phenotype tend to be either ancestral or derived. They did find a high degree of differentiation for the ensemble of T2D loci in East Asians and sub-Saharan Africans, however, suggesting that these groups might have experienced natural selection at T2D loci. These analyses provide some evidence for natural selection, but the validity of the thrifty genotype hypothesis remains largely unclear.
Speak man [104] proposed the “drifty” genotype hypothesis as an alternative to the thrifty genotype hypothesis. He argued that the existence of obesity- or diabetes-related genetic variants is simply because of random genetic drift over millions of years, as there is little evidence that humans were under strong selective pressure by famine throughout evolutionary history. Whether obesity and T2D loci represent thrifty or drifty genotypes is yet to be determined. Currently identified loci only represent a small part of the genetic architecture of obesity and T2D, and the causal genes or causal variants are largely unknown. Identification of more loci and more casual variants through ongoing fine-mapping efforts and whole-genome sequencing analyses will further illuminate the genetic basis of T2D in different ethnic groups.
Conclusion and Future Perspective
Significant progress in understanding the genetic determinants of T2D has been made through multiple waves of GWAS over a relatively short time. Although a majority of established T2D susceptibility loci were identified in European populations, recent GWAS in Asians have also made important contributions to the identification of susceptibility genes. These studies not only confirmed T2D susceptibility loci initially identified in European populations, but also identified novel susceptibility loci that provide new insights into the pathophysiology of diseases. At this juncture, however, currently known genetic variants do not appear to explain the excess susceptibility to T2D in Asians compared to European populations.
Several possibilities may explain this phenomenon. The casual genes/variants at known T2D loci, and their biological functions, are largely unclearly. Further studies with fine-mapping and sequencing analysis, and functional experiments, are needed to identify the causal genes/variants and mechanisms underlying the associations. Clearly, more genetic loci associated with T2D remain to be discovered since the currently known loci explained only a fraction of the heritability for T2D. More studies with large metaanalysis of GWAS, especially in Asian populations, are needed to identify loci that might be specific to Asians. Multi ethnic metaanalysis would be helpful to identify new loci and population-specific loci for T2D. In addition, rare (low-frequency) variants with larger effect size might account for some of the un explained heritability. Thus, deep-sequencing of the exomes or the whole genomes is needed. It is widely acknowledged that T2D is a result of the interplay between genetic and environmental factors. Investigation of gene–environment interactions is necessary to improve our understanding of the underlying pathophysiology of disease. Without consideration of environmental factors, current GWAS methods might miss important genetic variants specific to subgroups of the population as defined by some environmental exposures. Thus, the approach of genome-wide gene-environment interactions has the potential to uncover new T2D loci. These studies would help us better understand differences in T2D risk across ethnic groups.
Abbreviations and full names of genes
Abbreviations |
Full Names |
ANK1 |
Ankyrin 1, erythrocytic |
AP3S2 |
Adaptor-related protein complex 3, sigma 2 subunit |
ARL15 |
ADP-ribosylation factor-like 15 |
AMY1 |
Amylase, alpha 1 |
CDKAL1 |
CDK5 regulatory subunit associated protein 1-like 1 |
CDKN2A/B |
Cyclin-dependent kinase inhibitor 2A |
C2CD4A/B |
C2 calcium-dependent domain containing 4B |
FTO |
Fat mass and obesity associated |
FAF1 |
Fas (TNFRSF6) associated factor 1 |
GCC1/PAX4 |
GRIP and coiled-coil domain containing 1/ paired box 4 |
GLIS3 |
GLIS family zinc finger 3 |
GPSM1 |
G-protein signaling modulator 1 |
GRK5 |
G protein-coupled receptor kinase 5 |
HNF4A |
Hepatocyte nuclear factor 4, alpha |
HMG20A |
High mobility group 20A |
HHEX/IDE |
Hematopoietically expressed homeobox/ insulin-degrading enzyme |
KCNK16 |
Potassium channel, subfamily K, member 16 |
KCNQ1 |
Potassium voltage-gated channel, KQT-like subfamily, member 1 |
KCNJ11 |
Potassium inwardly-rectifying channel, subfamily J, member 11 |
KLF14 |
Kruppel-like factor 14 |
LPP |
LIM domain containing preferred translocation partner in lipoma |
MAEA |
Macrophage erythroblast attacher |
MIR129-LEP |
Mir129-leptin |
MPHOSPH9 |
M-phase phosphoprotein 9 |
PPARG |
Peroxisome proliferator-activated receptor gamma |
PTPRD |
Protein tyrosine phosphatase, receptor type, D |
PSMD6 |
Proteasome (prosome, macropain) 26S subunit, non-atpase, 6 |
PEPD |
Peptidase D |
PAX4 |
Paired box 4 |
POU5F1/
TCF19 |
POU class 5 homeobox 1/ transcription factor 19 |
RASGRP1 |
RAS guanyl releasing protein 1 (calcium and DAG-regulated) |
SRR |
Serine racemase |
SPRY2 |
Sprouty homolog 2 (Drosophila) |
SLC16A13 |
Solute carrier family 16, member 13 |
ST6GAL1 |
ST6 beta-galactosamide alpha-2,6-sialyltranferase 1 |
SGCG |
Sarcoglycan, gamma (35kda dystrophin-associated glycoprotein) |
SSR1/RREB1 |
Signal sequence receptor, alpha/ ras responsive element binding protein 1 |
TCF7L2 |
Transcription factor 7-like 2 |
TMEM163 |
Transmembrane protein 163 |
TMEM154 |
Transmembrane protein 154 |
UBE2E2 |
Ubiquitin-conjugating enzyme E2E 2 |
VPS26A |
Vacuolar protein sorting 26 homolog A |
ZFAND3 |
Zinc finger, AN1-type domain 3 |
Acknowledgements
Tao Wang is support by NIH grants 1R21HL118637 from the NHBLI. Xueyin Wang is supported by the China Scholarship Council.
References
- Wild S, Roglic G, Green A, Sicree R, King H (2004) Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes care 27: 1047-1053.
- Hu FB (2011) Globalization of diabetes: the role of diet, lifestyle, and genes. Diabetes care 34: 1249-1257.
- Chan J, So W, Ko G, Tong P, Yang X, et al. (2009) The Joint Asia Diabetes Evaluation (JADE) Program: a web-based program to translate evidence to clinical practice in Type 2 diabetes. Diabetic medicine : a journal of the British Diabetic Association 26: 693-699.
- Yang W, Lu J, Weng J, Jia W, Ji L, et al. (2010) Prevalence of diabetes among men and women in China. The New England journal of medicine 362: 1090-1101.
- Rankinen T, Zuberi A, Chagnon YC, Weisnagel SJ, Argyropoulos G, et al. (2006) The human obesity gene map: the 2005 update. Obesity (Silver Spring, Md.) 14: 529-644.
- Saunders CL, Chiodini BD, Sham P, Lewis CM, Abkevich V, et al. (2007) Meta-analysis of genome-wide linkage studies in BMI and obesity. Obesity (Silver Spring, Md.) 15: 2263-2275.
- Altshuler D, Hirschhorn JN, Klannemark M, Lindgren CM, Vohl MC, et al. (2000) The common PPARgamma Pro12Ala polymorphism is associated with decreased risk of type 2 diabetes. Nature genetics 26: 76-80.
- Gloyn AL, Weedon MN, Owen KR, Turner MJ, Knight BA, et al. (2003) Large-scale association studies of variants in genes encoding the pancreatic beta-cell KATP channel subunits Kir6.2 (KCNJ11) and SUR1 (ABCC8) confirm that the KCNJ11 E23K variant is associated with type 2 diabetes. Diabetes 52: 568-572.
- Gouda HN, Sagoo GS, Harding AH, Yates J, Sandhu MS, et al. (2010) The association between the peroxisome proliferator-activated receptor-gamma2 (PPARG2) Pro12Ala gene variant and type 2 diabetes mellitus: a HuGE review and meta-analysis. American journal of epidemiology 171: 645-655.
- Ludovico O, Pellegrini F, Di Paola R, Minenna A, Mastroianno S, et al. (2007) Heterogeneous effect of peroxisome proliferator-activated receptor gamma2 Ala12 variant on type 2 diabetes risk. Obesity (Silver Spring, Md.) 15: 1076-1081.
- Yang L, Zhou X, Luo Y, Sun X, Tang Y, et al. (2012) Association between KCNJ11 gene polymorphisms and risk of type 2 diabetes mellitus in East Asian populations: a meta-analysis in 42,573 individuals. Molecular biology reports 39: 645-659.
- Grant SF, Thorleifsson G, Reynisdottir I, Benediktsson R, Manolescu A, et al. (2006) Variant of transcription factor 7-like 2 (TCF7L2) gene confers risk of type 2 diabetes. Nature genetics 38: 320-323.
- Saxena R, Voight BF, Lyssenko V, Burtt NP, de Bakker PI, et al. (2007) Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science (New York, N.Y.) 316: 1331-1336.
- Scott LJ, Mohlke KL, Bonnycastle LL, Willer CJ, Li Y, et al. (2007) A genome- wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science (New York, N.Y.) 316: 1341-1345.
- Zeggini E, Weedon MN, Lindgren CM, Frayling TM, Elliott KS, et al. (2007) Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science (New York, N.Y.) 316: 1336-1341.
- Luo Y, Wang H, Han X, Ren Q, Wang F, et al. (2009) Meta-analysis of the association between SNPs in TCF7L2 and type 2 diabetes in East Asian population. Diabetes research and clinical practice 85: 139-146.
- Tong Y, Lin Y, Zhang Y, Yang J, Zhang Y, et al. (2009) Association between TCF7L2 gene polymorphisms and susceptibility to type 2 diabetes mellitus: a large Human Genome Epidemiology (HuGE) review and meta-analysis. BMC medical genetics 10: 15.
- Chang YC, Chang TJ, Jiang YD, Kuo SS, Lee KC, et al. (2007) Association study of the genetic polymorphisms of the transcription factor 7-like 2 (TCF7L2) gene and type 2 diabetes in the Chinese population. Diabetes 56: 2631-2637.
- Ng MC, Tam CH, Lam VK, So WY, Ma RC, et al. (2007) Replication and identification of novel variants at TCF7L2 associated with type 2 diabetes in Hong Kong Chinese. The Journal of clinical endocrinology and metabolism: 92: 3733-3737.
- (2007) Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 447: 661-678.
- Steinthorsdottir V, Thorleifsson G, Reynisdottir I, Benediktsson R, Jonsdottir T, et al. (2007) A variant in CDKAL1 influences insulin response and risk of type 2 diabetes. Nature genetics 39: 770-775.
- Sladek R, Rocheleau G, Rung J, Dina C, Shen L, et al. (2007) A genomewide association study identifies novel risk loci for type 2 diabetes. Nature 445: 881-885.
- Sandhu MS, Weedon MN, Fawcett KA, Wasson J, Debenham SL, et al. (2007) Common variants in WFS1 confer risk of type 2 diabetes. Nature genetics 39: 951-953.
- Gudmundsson J, Sulem P, Steinthorsdottir V, Bergthorsson JT, Thorleifsson G, et al. (2007) Two variants on chromosome 17 confer prostate cancer risk, and the one in TCF2 protects against type 2 diabetes. Nature genetics 39: 977-983.
- Zeggini E, Scott LJ, Saxena R, Voight BF, Marchini JL, et al. (2008) Metaanalysis of genome-wide association data and large-scale replication identifies additional susceptibility loci for type 2 diabetes. Nature genetics 40: 638-645.
- Prokopenko I, Langenberg C, Florez JC, Saxena R, Soranzo N, et al. (2009) Variants in MTNR1B influence fasting glucose levels. Nature genetics 41:77-81.
- Lyssenko V, Nagorny CL, Erdos MR, Wierup N, Jonsson A, et al. (2009) Common variant in MTNR1B associated with increased risk of type 2 diabetes and impaired early insulin secretion. Nature genetics 41: 82-88.
- Bouatia-Naji N, Bonnefond A, Cavalcanti-Proenca C, Sparso T, Holmkvist J, et al. (2009) A variant near MTNR1B is associated with increased fasting plasma glucose levels and type 2 diabetes risk. Nature genetics 41: 89-94.
- Rung J, Cauchi S, Albrechtsen A, Shen L, Rocheleau G, et al. (2009) Genetic variant near IRS1 is associated with type 2 diabetes, insulin resistance and hyperinsulinemia. Nature genetics 41: 1110-1115.
- Dupuis J, Langenberg C, Prokopenko I, Saxena R, Soranzo N, et al. (2010) New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nature genetics 42: 105-116.
- Voight BF, Scott LJ, Steinthorsdottir V, Morris AP, Dina C, et al. (2010) Twelve type 2 diabetes susceptibility loci identified through large-scale association analysis. Nature genetics 42: 579-589.
- Qi L, Cornelis MC, Kraft P, Stanya KJ, Linda Kao WH, et al. (2010) Genetic variants at 2q24 are associated with susceptibility to type 2 diabetes. Human molecular genetics 19: 2706-2715.
- Yasuda K, Miyake K, Horikawa Y, Hara K, Osawa H, et al. (2008) Variants in KCNQ1 are associated with susceptibility to type 2 diabetes mellitus. Nature genetics 40: 1092-1097.
- Unoki H, Takahashi A, Kawaguchi T, Hara K, Horikoshi M, et al. (2008) SNPs in KCNQ1 are associated with susceptibility to type 2 diabetes in East Asian and European populations. Nature genetics 40: 1098-1102.
- Tsai FJ, Yang CF, Chen CC, Chuang LM, Lu CH, et al. (2010) A genome-wide association study identifies susceptibility variants for type 2 diabetes in Han Chinese. PLoS genetics 6: e1000847.
- Shu XO, Long J, Cai Q, Qi L, Xiang YB, et al. (2010) Identification of new genetic risk variants for type 2 diabetes. PLoS Genet 6: e1001127.
- Yamauchi T, Hara K, Maeda S, Yasuda K, Takahashi A, et al. (2010) A genome-wide association study in the Japanese population identifies susceptibility loci for type 2 diabetes at UBE2E2 and C2CD4A-C2CD4B. Nature genetics 42: 864-868.
- Cho YS, Chen CH, Hu C, Long J, Ong RT, et al. (2012) Meta-analysis of genome-wide association studies identifies eight new loci for type 2 diabetes in east Asians. Nature genetics 44: 67-72.
- Imamura M, Maeda S, Yamauchi T, Hara K, Yasuda K, et al. (2012) A singlenucleotide polymorphism in ANK1 is associated with susceptibility to type 2 diabetes in Japanese populations. Human molecular genetics 21: 3042-3049.
- Li H, Gan W, Lu L, Dong X, Han X, et al. (2013) A genome-wide association study identifies GRK5 and RASGRP1 as type 2 diabetes loci in Chinese Hans. Diabetes 62: 291-298.
- Ma RC, Hu C, Tam CH, Zhang R, Kwan P, et al. (2013) Genome-wide association study in a Chinese population identifies a susceptibility locus for type 2 diabetes at 7q32 near PAX4. Diabetologia 56: 1291-1305.
- Hara K, Fujita H, Johnson TA, Yamauchi T, Yasuda K, et al. (2014) Genome- wide association study identifies three novel loci for type 2 diabetes. Human molecular genetics 23: 239-246.
- Kooner JS, Saleheen D, Sim X, Sehmi J, Zhang W, et al. (2011) Genomewide association study in individuals of South Asian ancestry identifies six new type 2 diabetes susceptibility loci. Nature genetics 43: 984-989.
- Saxena R, Saleheen D, Been LF, Garavito ML, Braun T, et al. (2013) Genome- wide association study identifies a novel locus contributing to type 2 diabetes susceptibility in Sikhs of Punjabi origin from India. Diabetes 62: 1746-1755.
- Tabassum R, Chauhan G, Dwivedi OP, Mahajan A, Jaiswal A, et al. (2013) Genome-wide association study for type 2 diabetes in Indians identifies a new susceptibility locus at 2q21. Diabetes 62: 977-986.
- Barhanin J, Lesage F, Guillemare E, Fink M, Lazdunski M, et al. (1996) K(V)LQT1 and lsK (minK) proteins associate to form the I(Ks) cardiac potassium current. Nature 384: 78-80.
- Ullrich S, Su J, Ranta F, Wittekindt OH, Ris F, et al. (2005) Effects of I(Ks) channel inhibitors in insulin-secreting INS-1 cells. Pflugers Archiv : European journal of physiology 451: 428-436.
- Hu C, Wang C, Zhang R, Ma X, Wang J, et al. (2009) Variations in KCNQ1 are associated with type 2 diabetes and beta cell function in a Chinese population. Diabetologia 52: 1322-1325.
- Qi Q, Li H, Loos RJ, Liu C, Wu Y, et al. (2009) Common variants in KCNQ1 are associated with type 2 diabetes and impaired fasting glucose in a Chinese Han population. Hum Mol Genet 18: 3508-3515.
- Tan JT, Nurbaya S, Gardner D, Ye S, Tai ES, et al. (2009) Genetic variation in KCNQ1 associates with fasting glucose and beta-cell function: a study of 3,734 subjects comprising three ethnicities living in Singapore. Diabetes 58: 1445-1449.
- Chagnon MJ, Uetani N,Tremblay ML (2004) Functional significance of the LAR receptor protein tyrosine phosphatase family in development and diseases. Biochemistry and cell biology = Biochimie et biologie cellulaire 82: 664-675.
- Inagaki N, Kuromi H, Gonoi T, Okamoto Y, Ishida H, et al. (1995) Expression and role of ionotropic glutamate receptors in pancreatic islet cells. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 9: 686-691.
- Jaggi F, Cabrita MA, Perl AK,Christofori G (2008) Modulation of endocrine pancreas development but not beta-cell carcinogenesis by Sprouty4. Molecular cancer research : MCR, 6, 468-482.
- Hartley T, Brumell J, Volchuk A (2009) Emerging roles for the ubiquitinproteasome system and autophagy in pancreatic beta-cells. American journal of physiology. Endocrinology and metabolism 296: E1-10.
- Warton K, Foster NC, Gold WA, Stanley KK (2004) A novel gene family induced by acute inflammation in endothelial cells. Gene 342: 85-95.
- Yuan J, Li T, Yin XB, Guo L, Jiang X, et al. (2006) Characterization of prolidase activity using capillary electrophoresis with tris(2,2'-bipyridyl) ruthenium(II) electrochemiluminescence detection and application to evaluate collagen degradation in diabetes mellitus. Analytical chemistry 78: 2934-2938.
- Takeuchi F, Serizawa M, Yamamoto K, Fujisawa T, Nakashima E, et al. (2009) Confirmation of multiple risk Loci and genetic impacts by a genomewide association study of type 2 diabetes in the Japanese population. Diabetes 58: 1690-1699.
- Silander K, Mohlke KL, Scott LJ, Peck EC, Hollstein P, et al. (2004) Genetic variation near the hepatocyte nuclear factor-4 alpha gene predicts susceptibility to type 2 diabetes. Diabetes 53: 1141-1149.
- Barroso I, Luan J, Wheeler E, Whittaker P, Wasson J, et al. (2008) Population- specific risk of type 2 diabetes conferred by HNF4A P2 promoter variants: a lesson for replication studies. Diabetes 57: 3161-3165.
- Fajans SS, Bell GI, Polonsky KS (2001) Molecular mechanisms and clinical pathophysiology of maturity-onset diabetes of the young. The New England journal of medicine 345: 971-980.
- Kang HS, Kim YS, ZeRuth G, Beak JY, Gerrish K, et al. (2009) Transcription factor Glis3, a novel critical player in the regulation of pancreatic betacell development and insulin gene expression. Molecular and cellular biology 29: 6366-6379.
- Girard C, Duprat F, Terrenoire C, Tinel N, Fosset M, et al. (2001) Genomic and functional characteristics of novel human pancreatic 2P domain K(+) channels. Biochemical and biophysical research communications 282: 249- 256.
- Soni S, Bala S, Gwynn B, Sahr KE, Peters LL, et al. (2006) Absence of erythroblast macrophage protein (Emp) leads to failure of erythroblast nuclear extrusion. The Journal of biological chemistry 281: 20181-20189.
- Luke MR, Houghton F, Perugini MA,Gleeson PA (2005) The trans-Golgi network GRIP-domain proteins form alpha-helical homodimers. The Biochemical journal 388: 835-841.
- Brun T, Gauthier BR (2008) A focus on the role of Pax4 in mature pancreatic islet beta-cell expansion and survival in health and disease. Journal of molecular endocrinology 40: 37-45.
- Wang X, Chen CF, Baker PR, Chen PL, Kaiser P, et al. (2007) Mass spectrometric characterization of the affinity-purified human 26S proteasome complex. Biochemistry 46: 3553-3565.
- Bennett V, Baines AJ (2001) Spectrin and ankyrin-based pathways: metazoan inventions for integrating cells into tissues. Physiological reviews 81: 1353-1392.
- Soranzo N, Sanna S, Wheeler E, Gieger C, Radke D, et al. (2010) Common variants at 10 genomic loci influence hemoglobin A(1)(C) levels via glycemic and nonglycemic pathways. Diabetes 59: 3229-3239.
- Krilov L, Nguyen A, Miyazaki T, Unson CG, Williams R, et al. (2011) Dual mode of glucagon receptor internalization: role of PKCalpha, GRKs and beta-arrestins. Experimental cell research: 317: 2981-2994.
- Tran TM, Jorgensen R, Clark RB (2007) Phosphorylation of the beta2- adrenergic receptor in plasma membranes by intrinsic GRK5. Biochemistry 46: 14438-14449.
- Seibold A, January BG, Friedman J, Hipkin RW, Clark RB (1998) Desensitization of beta2-adrenergic receptors with mutations of the proposed G protein-coupled receptor kinase phosphorylation sites. The Journal of biological chemistry 273: 7637-7642.
- Barker BL, Benovic JL (2011) G protein-coupled receptor kinase 5 phosphorylation of hip regulates internalization of the chemokine receptor CXCR4. Biochemistry 50: 6933-6941.
- Parameswaran N, Pao CS, Leonhard KS, Kang DS, Kratz M, et al. (2006) Arrestin-2 and G protein-coupled receptor kinase 5 interact with NFkappaB1 p105 and negatively regulate lipopolysaccharide-stimulated ERK1/2 activation in macrophages. The Journal of biological chemistry 281: 34159-34170.
- Brooks-Worrell B, Palmer JP (2012) Immunology in the Clinic Review Series; focus on metabolic diseases: development of islet autoimmune disease in type 2 diabetes patients: potential sequelae of chronic inflammation. Clinical and experimental immunology 167: 40-46.
- Habener JF, Kemp DM ,Thomas MK (2005) Minireview: transcriptional regulation in pancreatic development. Endocrinology 146: 1025-1034.
- Tokuyama Y, Matsui K, Ishizuka T, Egashira T,Kanatsuka A (2006) The Arg121Trp variant in PAX4 gene is associated with beta-cell dysfunction in Japanese subjects with type 2 diabetes mellitus. Metabolism: clinical and experimental 55: 213-216.
- Plengvidhya N, Kooptiwut S, Songtawee N, Doi A, Furuta H, et al. (2007) PAX4 mutations in Thais with maturity onset diabetes of the young. The Journal of clinical endocrinology and metabolism 92: 2821-2826.
- Maffei M, Halaas J, Ravussin E, Pratley RE, Lee GH, et al. (1995) Leptin levels in human and rodent: measurement of plasma leptin and ob RNA in obese and weight-reduced subjects. Nature medicine 1: 1155-1161.
- Blumer JB, Lord K, Saunders TL, Pacchioni A, Black C, et al. (2008) Activator of G protein signaling 3 null mice: I. Unexpected alterations in metabolic and cardiovascular function. Endocrinology 149: 3842-3849.
- Hirai T, Fukui Y, Motojima K (2007) PPARalpha agonists positively and negatively regulate the expression of several nutrient/drug transporters in mouse small intestine. Biological & pharmaceutical bulletin 30: 2185-2190.
- Morris AP, Voight BF, Teslovich TM, Ferreira T, Segre AV, et al. (2012) Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nature genetics 44: 981-990.
- Dufresne AM, Smith RJ (2005) The adapter protein GRB10 is an endogenous negative regulator of insulin-like growth factor signaling. Endocrinology 146: 4399-4409.
- Woodard-Grice AV, McBrayer AC, Wakefield JK, Zhuo Y, Bellis SL (2008) Proteolytic shedding of ST6Gal-I by BACE1 regulates the glycosylation and function of alpha4beta1 integrins. The Journal of biological chemistry 283:26364-26373.
- Groh S, Zong H, Goddeeris MM, Lebakken CS, Venzke D, et al. (2009) Sarcoglycan complex: implications for metabolic defects in muscular dystrophies. The Journal of biological chemistry 284: 19178-19182.
- Sim X, Ong RT, Suo C, Tay WT, Liu J, et al. (2011) Transferability of Type 2 Diabetes Implicated Loci in Multi-Ethnic Cohorts from Southeast Asia. PLoS Genetics 7: e1001363.
- Waters KM, Stram DO, Hassanein MT, Le Marchand L, Wilkens LR, et al. (2010) Consistent association of type 2 diabetes risk variants found in Europeans in diverse racial and ethnic groups. PLoS genetics 6: e1001078
- Wu Y, Li H, Loos RJ, Yu Z, Ye X, et al. (2008) Common variants in CDKAL1, CDKN2A/B, IGF2BP2, SLC30A8, and HHEX/IDE genes are associated with type 2 diabetes and impaired fasting glucose in a Chinese Han population. Diabetes 57: 2834-2842.
- Meigs JB, Shrader P, Sullivan LM, McAteer JB, Fox CS, et al. (2008) Genotype score in addition to common risk factors for prediction of type 2 diabetes. The New England journal of medicine 359: 2208-2219.
- Lyssenko V, Jonsson A, Almgren P, Pulizzi N, Isomaa B, et al. (2008) Clinical risk factors, DNA variants, and the development of type 2 diabetes. The New England journal of medicine 359: 2220-2232.
- van Hoek M, Dehghan A, Witteman JC, van Duijn CM, Uitterlinden AG, et al. (2008) Predicting type 2 diabetes based on polymorphisms from genomewide association studies: a population-based study. Diabetes 57: 3122-3128.
- Lango H, Palmer CN, Morris AD, Zeggini E, Hattersley AT, et al. (2008) Assessing the combined impact of 18 common genetic variants of modest effect sizes on type 2 diabetes risk. Diabetes 57: 3129-3135.
- Cornelis MC, Qi L, Zhang C, Kraft P, Manson J, et al. (2009) Joint effects of common genetic variants on the risk for type 2 diabetes in U.S. men and women of European ancestry. Annals of internal medicine 150: 541-550.
- Sparso T, Grarup N, Andreasen C, Albrechtsen A, Holmkvist J, et al. (2009) Combined analysis of 19 common validated type 2 diabetes susceptibility gene variants shows moderate discriminative value and no evidence of genegene interaction. Diabetologia 52: 1308-1314.
- Qi Q, Li H, Wu Y, Liu C, Wu H, et al. (2010) Combined effects of 17 common genetic variants on type 2 diabetes risk in a Han Chinese population. Diabetologia 53: 2163-2166.
- Janipalli CS, Kumar MV, Vinay DG, Sandeep MN, Bhaskar S, et al. (2012) Analysis of 32 common susceptibility genetic variants and their combined effect in predicting risk of Type 2 diabetes and related traits in Indians. Diabetic medicine : a journal of the British Diabetic Association 29: 121-127.
- Mahajan A, Go MJ, Zhang W, Below JE, Gaulton KJ, et al. (2014) Genomewide trans-ancestry meta-analysis provides insight into the genetic architecture of type 2 diabetes susceptibility. Nature genetics 46 : 234-244.
- Willer CJ, Speliotes EK, Loos RJ, Li S, Lindgren CM, et al. (2009) Six new loci associated with body mass index highlight a neuronal influence on body weight regulation. Nature genetics 41: 25-34.
- Craddock N, Hurles ME, Cardin N, Pearson RD, Plagnol V, et al. (2010) Genome-wide association study of CNVs in 16,000 cases of eight common diseases and 3,000 shared controls. Nature 464: 713-720.
- Bae JS, Cheong HS, Kim JH, Park BL, Kim JH, et al. (2011) The genetic effect of copy number variations on the risk of type 2 diabetes in a Korean population. PloS one 6: e19091.
- Perry GH, Dominy NJ, Claw KG, Lee AS, Fiegler H, et al. (2007) Diet and the evolution of human amylase gene copy number variation. Nature genetics 39: 1256-1260.
- Neel JV (1962) Diabetes mellitus: a "thrifty" genotype rendered detrimental by "progress"? American journal of human genetics 14: 353-362.
- Southam L, Soranzo N, Montgomery SB, Frayling TM, McCarthy MI, et al. (2009) Is the thrifty genotype hypothesis supported by evidence based on confirmed type 2 diabetes- and obesity-susceptibility variants? Diabetologia 52: 1846-1851.
- Klimentidis YC, Abrams M, Wang J, Fernandez JR, Allison DB (2011) Natural selection at genomic regions associated with obesity and type-2 diabetes: East Asians and sub-Saharan Africans exhibit high levels of differentiation at type-2 diabetes regions. Human genetics 129: 407-418.
- Speakman JR (2008) Thrifty genes for obesity, an attractive but flawed idea, and an alternative perspective: the 'drifty gene' hypothesis. International journal of obesity 32: 1611-1617.





