Intra-maxillary Molecular Releasing and its Application in the Assistance of Neurodegenerative Disease Therapeutics
Li Y J
Department of Nursing, St. Mary’s Junior College of Medicine, Nursing, and Management, Yilan 266, Taiwan.
*Corresponding Author
Yu-Jung Li
Department of Nursing, St. Mary’s Junior College of Medicine,
Nursing, and Management, Yilan 266, Taiwan.
Tel.: +886 2 2795-6030/+886-980-023203
E-mail: richard513.tw@yahoo.com.tw
Received: February 29, 2016; Accepted: March 21, 2016; Published: March 31, 2016
Citation: Li Y J (2016) Intra-maxillary Molecular Releasing and its Application in the Assistance of Neurodegenerative Disease Therapeutics. Int J Clin Ther Diagn. 4(1), 100-109.DOI : dx.doi.org/10.19070/2332-2926-1600021
Copyright: Li Y J© 2016. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
Abstract
Neurodegenerative disease (ND) is a complex concept involves multiple pathogenesis including neuronal demyelination, apoptosis under oxidative stress, neuroinflammation with further cortical and spinal atrophy, which may lead to cognition impairment and multiple neural dysfunctions. It may present as a chronic and irreversibly progressive disorder and shows the common pathological pictures among its various types including Alzheimer’s disease (AD), Parkinson’s disease (AD), Huntington’s disease (HD), multiple sclerosis (MS), and amyotrophic lateral sclerosis (ALS). The symptoms often occur among the elderly above 50 years old and last from years to decades. Such facts lead ND as the highest priority to geriatrics and its related medical care. Due to its chronic progressively characteristics, regularly invasive procedure arrangements may face the difficulty and regarded as unbearable in the clinical practice. Unfortunately, current available therapy is mainly on symptom relief and the potential therapeutic targets may focus on the macromolecules including oligomer, peptides, or even functional proteins with the quaternary structure since multiple proteinopathy involvement in ND pathogenesis. Such macromolecular administrations cause the challenges in the routine drug delivery selections due to the impossibility of the traditional oral intake.
Intra-maxillary molecular releasing is a newly drug delivery pathway via dental implant and may directly reach the inside bone marrow. It presents the advantages in long-term and continuous macromolecular releasing with relative painless process, which is appropriate for further applications in ND therapeutics. The stability of dental implant inside the oral cavity has also been proved for decades among more than millions around the world. However, such delivery pathway is currently under development and further interdisciplinary investigations are needed including therapeutics, pharmacokinetics, neurology, molecular biology, clinical dentistry, chemical engineering, automatics and further complementary metal-oxidesemiconductor (CMOS) technology to ensure the efficiency and prove the safety concern.
2.Introduction
3.Alzheimer’s disease (AD)
4.Parkinson disease (PD)
5.Huntington’s disease (HD)
6.Multiple Sclerosis (MS)
7.Amyotrophic lateral sclerosis (ALS)
8.Therapeutic developments of ND
9.Oxidative stress modification
10.Antibody therapy
11.Neuroregenerative therapy
12.Intra-maxillary molecular releasing in applications of ND therapeutics
13.Pharmacokinetics in intra-maxillary molecular releasing
14.Conclusion
15.Acknowledgments
16.References
Keywords
Neurodegenerative Disease (ND); Alzheimer’s Disease (AD); Parkinson’s Disease (AD); Huntington’s Disease (HD); Multiple Sclerosis (MS); Amyotrophic Lateral Sclerosis (ALS); Dental Implant; Intra-Maxillary Molecular Releasing; Complementary Metal-Oxide-Semiconductor (CMOS) Technology.
Introduction
Neurodegenerative disease (ND) is a special and complex concept presenting much different from other systematic diseases including infection, immune, nutrition insufficiency, hypoxia, etc. It may clinically present as a disorder or disability with partial congenital relationships, and involve inflammatory patterns including lymphocyte infiltration in pathological findings with metabolic origins. In the cellular point of view, neurons cannot exist alone and need surrounding supports including glial cells and blood supply. Furthermore, the neural system is a complete network, and some specific pathological changes may result in the next or previous connections, which may increase difficulties in the primary and secondary disease determinations. Some neurodegenerative diseases involve in cortex, basal ganglia, cerebellum, or spinal cord, while others may occur in the intermediate form or multi-systemic diseases. Despite of the complexity, most of the neurodegenerative diseases present in neuron decrease or even disappear with glial cell occupations in the later stages.
Alzheimer’s disease (AD)
AD is the most common neurodegenerative disorder among the elderly above fifty years old [1]. It affects 10% of the elderly above 65 years old, and around half of those over 85 years old [2, 3]. The typical symptoms include memory loss, cognitive impairment, and behavior dysfunctions. It may also induce secondary physiological dysfunctions including swallowing, and psychological involvement such as depression [4, 5]. The etiology of AD remains unclear due to its complex and multifactorial pathogenesis including genetic mutation [6, 7], abnormal protein procession, deposition, and aggregations [8, 9], inflammation and related immune responses [10-12], neurotrophic factors [13, 14], metabolic disorders [15-17], oxidative stress [18, 19], excitotoxicity [20, 21], and neurotransmitter maladjustment [22, 23].
In the pathological point of view, it may involve hippocampus and cortex regions with the following patterns:
- Brain atrophy: With the neuron disappear, the brain weight may decrease and the sulcus is widening with ventricle enlargement.
- Senile plaque: It may occur in the cortex and hippocampus, or even found in the basal ganglia and brain stem. In hematoxylin and eosin (H E) stain histology, it represents in eosinophilic round shape sized about 50 μm within an amyloid center [24]. The amyloid is mainly from the misfolded amyloid β proteins (Aβ), which is a 39-43 amino acid peptide with β-sheet conformation leading to neurodegeneration [25]. Various Aβ isoforms are determined including Aβ39-Aβ43, while Aβ40 and Aβ42 are the most common peptides inside the neurotoxic soluble oligomers or insoluble fibrils arising neuron damages [26-30].
- Neurofibrillary tangle (NFT): NFT is composed by the paired helical filaments (PHF) with 7-9 diameter twisted each other and forms a tangle within 80 μm [31, 32]. The PHF contains neurofilament tripeptide, neurotubules, Aβ, and tau protein. Tau protein is a microtubule-associated protein promoting its assembly to maintain axonal dynamics in a neuron [33, 34]. Currently NFT has been the key characteristics in pathology for AD diagnosis [35]. However, tau and NFT may be found not only in AD but among various ND and regarded as tauopathies [36]. It may be responsible for the primary origin to induce neurodegeneration generally, while it is regarded as the secondary event following amyloidogenesis in AD [37].
The above pathological findings may plot the characteristics of AD. Furthermore in the molecular level, NFT may have neural toxicity by reducing microtubule stability due to tau protein mutation, hyperphosphorylation, and aggregations [38, 39]. The human tau gene locates on chromosome 17 with 16 exons and produces six isoforms by different ways of splicing within two inserts closed to the N-terminus, and three to four highly conserved, imperfect repeat sequences microtubule-binding domains in the C-terminus [40, 41]. Such splicing produces two groups of tau isoforms including three (3R) or four (4R) microtubule-binding repeats. In normal human brain, the 3R:4R tau ratio is around 1:1, while it may increase during neurodegenerative tauopathies [42]. There are two major mutations that will result in tau aggregations into NFTs including intronic and coding mutations [43, 44]. Besides, tau phosphorylation is important in its microtubule-binding process, and insoluble, highly phosphorylated tau may arouse tauopathy and leads to neuronal loss [45]. That implies inhibition of protein kinases by targeting tau phosphorylation process may provide the therapeutic approach to reduce tau aggregations [46].
Current therapeutics of AD is mainly for progression decelerated. Acetylcholinesterase (AChE) inhibitors may provide cognitive enhancement. Donepezil, galantamine and rivastigmine may enhance cholinergic neurotransmission for mild-to-moderate AD [47, 48], while N-methyl-D-aspartate (NMDA) receptor antagonist memantine is recommended for moderate-to-severe AD [49, 50].
Parkinson disease (PD)
PD is the second common ND and first described by James Parkinson [51]. It is a progressive neurological motor dysfunction involves extrapyramidal system, and leads to tremors with hands and jaws, muscle rigidity, disability of postural balance, difficulty in initiating willed movements as akinesia, and slowness of movement as bradykinesia [52, 53]. Many patients also suffer from cognitive impairment as PD progresses. The prevalence of PD is around 0.15% to 0.2% among the population, and 1% of the elders above 60 years old [54, 55].
The basal ganglia include the caudate nucleus, the putamen, the globus pallidus, and the subthalamic nucleus. Furthermore, the substantia nigra inside the midbrain structure also has the connection to the basal ganglia. The caudate and the putamen are responsible to receive the cortex input to the basal ganglia, which are also known as the striatum [56]. There are intrinsic and extrinsic fibers inside the basal ganglia, including the excitatory postsynaptic potential (EPSP) and the inhibitory postsynaptic potential (IPSP) types of the synaptic activations. The intrinsic fibers mainly belong to the Acetylcholine (Ach) and Gammaaminobutyric acid (GABA) type, while the most importantly extrinsic fibers are the Dopamine (DA) type from the substantia nigra. Besides, the basal ganglia also has other input fibers including the Ach fibers from the thalamus, the Serotonin (5-HT) IPSP fibers from the red nucleus, and the glutamate IPSP fibers from the cortex.
Basically PD is the neurodegeneration among the DA fibers from the substantia nigra and input to the striatum [57]. Besides basal ganglia, the locus ceruleus, dorsal motor nucleus of vagus, and even the sympathetic system may be also involved. The DA, Norepinephrine (NE), and 5-HT levels are significantly decreased among the damage area. Furthermore, the tyrosine hydroxylase as the key enzyme for DA synthesis is also deficient among the DA neurons. The pathology findings present the senile plaque, NFT, and Lewy bodies, which are the round-shaped eosinophilic granules with deeper-colored nucleus inside the neuron. The Lewy body is composed by the neurofilaments within 8-10 nm diameters in width, but it does not contain PHF.
The secondary PD may be caused by brain damages with various origins including long-term drug administrations and traumatic injuries. However, the etiology of the primary PD still remains unclear. It involves various environmental factors including oxidative stress, DA metabolism, mitochondrial dysfunction, neuroinflammation, but rare genetic mutations have been described [58-63]. The pathological processes leading to mitochondrial dysfunction among the DA neurons will result in the accumulation of reactive oxygen species (ROS) and increase the risk of oxidative stress, which may cause further damages to the related proteins, lipids and nucleic acids among the DA neurons [64-66]. Actually, the oxidative stress as the imbalance between the excessive free radicals and the intracellular antioxidant system is also extremely important in AD and other neurodegenerative pathogenesis [67, 68].
Currently there is not therapeutics available to decelerate the neurodegenerative progressions, and the medication is purely palliative. Therefore the therapeutic goal for PD is mainly on DA level enhancement, which may be achieved by the DA precursor, L-dihydroxyphenylalanine administrations. It may cross through the blood-brain barrier (BBB) and promote the remaining neurons in the substantia nigra for DA production. However, such therapy will become ineffective after all the DA neurons die in the later stage of PD [69].
Huntington’s disease (HD)
In contrast to PD, HD is an autosomal dominant inherited neurodegenerative disorder with progressive and inevitably fatal syndrome. The prevalence of HD is around 5 per 100,000 among the population [70]. It shares some similar clinical characteristics among ND including hyperkinesia, dyskinesia, dementia, cognitive impairment, and personality changes. However, the above symptoms would not appear until their adulthood. The typical sign of HD is chorea corresponding to spontaneous and meaningless movements among various parts of the body beyond control.
It is first described by the physician George Huntington in 1872, and the related gene is identified as IT15 at short arm of chromosome 4 in 1993 [71]. IT15 will elongate with the repetitive sequence of cytosine, adenosine, and guanine (CAG) [72]. Furthermore, the length of CAG repetition will have influence on HD onset determination [73]. The pathological findings of HD are mainly on the significant neuron loss in the caudate nucleus, putamen, and globus pallidus [74]. In addition, the cerebral cortex may also be included. The structural destructions of basal ganglia and its loss of output intrinsic fibers to the thalamus may induce movement disorders, including hyperkinesia and ballism as violent and flinging movement of extremities. Similar to other ND, HD may lead to further mitochondrial dysfunction and the related oxidative stress [75].
Multiple Sclerosis (MS)
Traditionally MS is regarded as an inherited autoimmune disease with cell-mediated delayed hypersensitivity reactions. Patients suffering from MS often belong to the types of human leukocyte antigen (HLA-typing) A3, B7 and DR2. BBB destructions with plasma cell hyperactivity and immunoglobulin G (IgG) overproduction outside the vessels are found around 40% to 50% MS patients in the early stage. In the acute MS known as Malburg type, the pathological findings present Balo’s concentric sclerosis at the cortex due to demyelinating [76], and Devic’s neuromyelitis optica at the spinal cord and the optical nerves. However, MS often presents chronic progressions with MS plaque sized from millimeters to centimeters due to myelin disappearance and glial cell proliferations. Recently MS is gradually regarded as a kind of ND due to ineffective response to the immunotherapies including immunosuppressive and immunomodulatory administrations [77]. Besides, MS also shows some similar pathogenesis pictures to ND. Recent pathological findings proved that T lymphocyte infiltration is still presented in the lesion with parenchymal loss [78-80]. In the molecular level, mitochondrial dysfunction due to inflammatory process is dominant in demyelination [81, 82]. Furthermore, kinase and phosphatase imbalance also result in cytoskeletal destructions [83]. The above facts may provide hints for further therapeutic development of both MS and ND.
Amyotrophic lateral sclerosis (ALS)
ALS, also known as Lou Gehrig’s disease, is a life-threatening ND involved in the upper and lower motor neurons over the spinal cord, brain stem and the related motor cortex [84]. The mean onset age is about 55 years old, and the incidence is around 1-2 per 100,000 per year with 3-5 years of survival after diagnosis [85, 86]. The pathological changes include neuronal apoptosis, fiber demyelination, and glial cell increases in the anterior and lateral corticospinal tract, which may lead to progressive paralysis. The etiology remains unclear, but 10% of ALS is familial and relates to the genetic mutation of antioxidant enzyme copper and zinc superoxide dismutase (SOD1) [87]. Both of the sporadic and familial ALS may result from neuronal damage due to oxidative stress with free radical involvements [88-91]. Current potential therapeutics may include antioxidant, neuroregeneration and stem cell therapies [19, 92, 93].
Therapeutic developments of ND
Currently there is no effective agent for ND patients and the commercially available drugs are mainly for palliative to decelerate the progressions. However, recent investigations provide indications for specific drug designs and therapeutic developments of ND throughout the pathological mechanism and tissue engineering principles.
Oxidative stress modification
Oxidative stress is one of the major subcellular mechanisms, which may produce free radicals, overwhelm the antioxidant system, and result in neuronal dysfunction and further axonal damage among various ND [94, 66]. It is often occur in mitochondrion among various subcellular organs and produce hydrogen peroxide due to oxygen consumption and adenosine triphosphate (ATP) generation [95]. The Coenzyme Q10 (2,3-dimethoxy-5-methyl- 6-decaprenil-1,4-benzoquinone), also regarded as CoQ10 or vitamin Q10, is an important antioxidant agent participating in various subcellular events [19]. It mainly accumulates in the inner membrane of mitochondrion and inhibits lipid peroxidation [96, 97]. Generally it may be obtained from daily diet, supplements, and absorbed in the small intestine. In addition, it may also be produced from tyrosine endogenously, especially in the organs with highly metabolic rate. A sixteen-month clinical study with 80 patients presents 44% less functional decline in PD patients after oral administration of CoQ101200 mg per day [98]. Muller also reported the positive results with moderate symptom improvements after 360 mg of CoQ10 daily administrations [99]. Furthermore, daily intake of CoQ10 also improves learning and memory deficits by suppressing the oxidative stress and elevating the ATP level [100]. With 360 mg per day of CoQ10 administration for 2-8 weeks, the lactate level in the occipital cortex is suppressed [101], but it may return to the baseline value after stop administration. CoQ10 is also considered as the potential therapeutics of ALS since the oxidative stress and mitochondrial dysfunction participate in ALS progressions [102].
Antibody therapy
Since complex physiological event involvements as proteinopathies among various types of ND progressions, selective protein targeting has been considered as the reasonable approach in ND therapeutic development [103]. However, safety concern for such systemic immunotherapy should be carefully evaluated before further clinical trial. The US Food and Drug Administration (FDA) has approved several intravenous immunoglobulin (IVIg) injections as safe for several immune disorders. Such therapy may provide anti-Aβ antibodies to suppress Aβ fibril formation and enhance the Aβ clearance from the brain to avoid its neuronal toxicity [104-108]. In addition, the anti-Aβ antibodies present the ability to cross the BBB and may directly reach the lesion in central neural system (CNS). Several pilot clinical trials also reported the positive results for regular anti-Aβ antibody injections [109- 111]. Tauopathy is also regarded as another important target for immunotherapy development. The mutative tau may arouse NFT and lead to further frontotemporal dementia with parkinsonism (FTDP). The mouse model shows positive results after anti-tau immunotherapy by motor performance improvement [112]. The detected antibody in the transgenic mice also shows its ability to cross the BBB.
In the contrary, targeting mHtt protein immunotherapy reaches the difficulty due to its intracellular localization [113]. As the above descriptions, the mutative gene may encode proteins which lead to conformational changes toward monomers, oligomers, protofibrils, and the related fibrillar structures including A βplaques, Lewy bodies, α-synuclein and mHtt aggregates among various ND. Such proteinopathies provide the possibility for intrabody therapy [114, 115]. The intrabodies (iAbs) are the intracellular fragments which may recognize the specific antigen domains and provide the potential in further AD, PD, and HD therapeutics [103]. However, the efficiency of these immunotherapies is not certain and further clinical trial arrangements are needed for both efficiency and safety confirm.
Neuroregenerative therapy
As the basic concept of ND including neuronal damage, apoptosis and the related progressive dysfunctions, the neuroregenerative therapy is considered base on the principle of tissue engineering, including neurorestoration as stem cell therapy, neurogenesis as neurotrophilins and growth factors, and neurotrophins [92, 93, 116-119]. The adult neural stem cell has been proved its existence in the 1990s [120, 121], and present the possibility of neuroregeneration in various ND models [122]. However, the mammalian central nervous system (CNS) still presents in a limited capacity of neurogenesis and proliferative property compared to other tissues. Most of the neural stem cells or related progenitors may reside in the subventricular zone (SVZ), the lateral ventricular wall, and the subgranular zone of the hippocampal dentate gyrus, which may provide the possibility for neurogenesis [123-125].
The natural occurring stem cells may be divided into two groups by their capacity of differentiation including the totipotent, or the pluripotent embryonic stem cells (ESC), and the multipotent stem cells, such as fetal stem cells (FSC) and the adult stem cells. The ESCs present unlimited proliferative and differentiation capacities [126]. However, the applications are highly restricted due to various ethical concerns. Due to the challenges in neural stem cell extraction and the difficulty in mesenchymal stem cell (MSC) transdifferentiation, techniques for induced pluripotent stem cell (iPS) producing are developed via various reprogramming processes with specific transcription factors, such as Oct4, Sox2, Klf4, and c-Myc [127]. However, such therapy may face the challenges in safety concern for iPS induced teratomas [128], efficiency limitation in continuous stem cell delivery, and the invasive procedures in clinical practice to reach the central venous system. The above restrictions may limit the stem cell therapy in the life-threatening ND applications including ALS and MS [129]. In contrast, a clinical trial was conducted for AD patients with directly iPS delivery into CNS in 2005 [116], and reported positive results by both cognitive scales and positron emission tomography (PET) scanning after 22 months’ follow-up.
Neurotrophins and the related cytokines, such as Nerve Growth Factor (NGF), Brain-derived Nerve Factor (BDNF), neurotrophin 3 (NT3), and neurotrophin 4 (NT4) also present the capacity to promote neuroregeneration in both normal CNS development and pathological aging [130-132]. However, these cytokines may systemically participate in various subcellular events throughout tropomyosin-related kinase (Trk) receptors and activate the relative signaling pathways. Safety concern with enough clinical trial evaluations are needed in such applications.
Intra-maxillary molecular releasing in applications of ND therapeutics
The pathological process of neurodegeneration presents as multiple cellular and subcellular event involvement, and that increases the difficulty on therapeutic development. Current ND therapies are majorly on symptom relief. The following situations should be taken into consideration in further therapeutic development:
- Most ND present as the chronic progressive dysfunction without life-threatening properties, and the process may last for decades. For the above reason, regularly invasive procedures may be unbearable in applications to the ND patients, such as daily injections to prevent from neurodegeneration. That also means repeated surgical approaches including CNS delivery in stem cell therapy may be unacceptable in such ND patients.
- Most of the sufferings belong to the elderly, so that geriatrics becomes important in therapeutic considerations including cross-interactions and the related side effects of other cardiovascular or antibiotic agents in ND drug delivery.
- The pathogenesis of ND present in the properties of proteinopathy including tauopathy, oxidative stress andneuroinflammation. That implies peptide, oligomer,and other functional macromolecular delivery may be unavoidable in further therapeutic developments. However, such macromolecular absorption present unsteady, or even seems impossible directly byoral intake passing through the gastrointestinal (GI) tract. In contrast, regularly intravenous injection of these macromolecules may become an invasive procedure with pain and irritations, which seems the paradox of the first point.
- The penetrative ability crossing through BBB may be much important in ND drug design considerations since the ability enhancement may lower down the relative side effects of ND administrations, especially in such chronic disease with the demand of long-term and regular dosage administrations.
Despite the above restrictions, we may consider the optimal drug concentration in plasma to meet the ND therapeutic demand. As the progressively functional disorder with metabolic and other intrinsic origins, the demand of ND curing dosage may present steady and continuous with a certain degree of concentration rather than the massive and rapid drug releasing types in specific infectious applications.
Intra-maxillary molecular releasing in applications of ND therapeutics
The development of dental implant has been lasting for decades and regarded as a mature procedure for tooth reconstructions since Dr. Brånemark announced the principle of osseointegration in the 1970’s [133]. The basic principle of dental implantology is shown in Figure 1(A). The tooth structure includes the outside enamel crown and the intra-bony root cementum, and its pain receptors are mainly from the pulp structure (a) inside the tooth, and the periodontal ligament (PDL) surrounding its outer surface and directly contact the bone marrow (b). Within dental implant placement after tooth loss, the above pain origins are both removed and offer a pathway for continuous molecular releasing and bio-sensing without painful sensations [134, 135]. Figure 1(B) presents the molecular releasing pathway from outside oral cavity passing through the titanium implant fixture to reach the inside bone marrow. Such pathway may provide some characteristics within the related advantages compared to the traditional drug delivery procedures as followings.
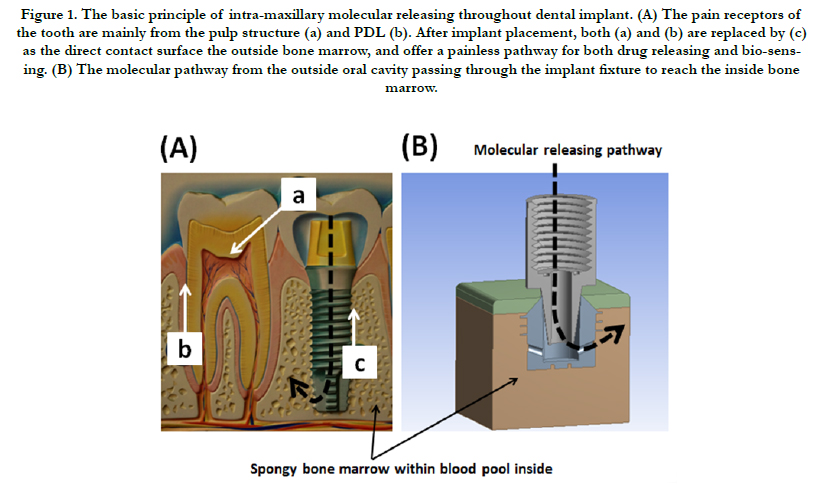
Figure 1. The basic principle of intra-maxillary molecular releasing throughout dental implant. (A) The pain receptors of the tooth are mainly from the pulp structure (a) and PDL (b). After implant placement, both (a) and (b) are replaced by (c) as the direct contact surface the outside bone marrow, and offer a painless pathway for both drug releasing and bio-sensing. (B) The molecular pathway from the outside oral cavity passing through the implant fixture to reach the inside bone marrow.
- Compared to other traditional drug delivery procedures, the obvious advantage may be relative painless and continuous drug releasing throughout the whole process by such intramaxillary pathway.
- Macromolecular or even cellular delivery with longterm and continuous property is possible via such intramaxillary pathway. Moreover, the surrounding bone marrow environments may increase the initial stem cell survival rate compared to the intravenous injection type due to the blood flow velocity. However, we should notice that stem cell delivery into the maxillary bone marrow may face the difficulty crossing through BBB in ND therapeutic applications.
- The intra-maxillary molecular releasing may avoid thrombolism and further risks of stroke occur due to its bone marrow structure compared to other continuous venous drug releasing.
- Multiple drug type delivery at the same time, which refers as combination therapy is also possible within this intramaxillary drug delivery pathway.
In contrast, it also presents some restrictions as following.
- To maintain the stability of the dental implant and avoid from further surrounding bony resorption, drug polymerization with slowly degraded properties to achieve molecular releasing is needed. That implies rapid and massive drug releasing is impossible throughout such drug delivery pathway. However, as multiple proteinopathy involvements including metabolic, neuroinflammation and other genetic mutative events, the ND therapeutics may require the continuous and steady drug releasing type rather than the rapid onset ones.
- The types of the loading drugs should be carefully evaluated to avoid further destructions of the osseointegrative implant structure. For example, the low pH value molecules with immuno-induced properties may be an inappropriate target for intra-maxillary releasing.
For further detail discussions in such intra-maxillary drug delivery pathway and its applications of ND therapeutics, we will review the principle of pharmacokinetics.
Pharmacokinetics in intra-maxillary molecular releasing
After administrations of the drug loading dosages via various pathways including oral intake, intravenous, intramuscular, subcutaneous, and so on, the concentration in plasma may gradually increase to reach the maximum value (Cmax), and then decay slowly due to the intrinsic eliminative process. Figure 2(A) plots the concentration curve throughout the whole administration process, and area under the curve corresponds to the bioavailability. Once when the concentration presents higher than the minimal effective concentration (MEC) regarded as onset of the loading drug, it will lead to further meaningful biological functions until the concentration lower than MEC. The period when the loading concentration presents higher than MEC may correspond to the drug duration. However, it should remain under the minimal toxic concentration (MTC) throughout the whole drug loading process. Otherwise the drug toxicities and other significant side effects may occur.
The drug design of the rapid onset or sustained releasing types may accords to the above principles, which shows in the Figure 2(B). The drug concentration of the rapid onset type may increase immediately higher than MEC after administration, but maintain in a relative short period and degrade soon. In contrast, the sustained type may present as slower onset time with longer period remaining above the MEC. However, since intra-maxillary drug releasing presents as the slowly polymeric dissolved type, its concentration in plasma may maintain in a certain level and last for a relative long period compared to other drug delivery pathways, which may present more benefits in further ND therapeutic applications.
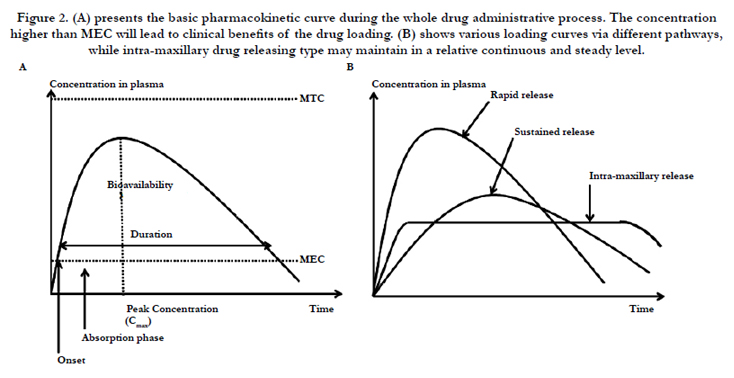

Figure 2. (A) presents the basic pharmacokinetic curve during the whole drug administrative process. The concentration higher than MEC will lead to clinical benefits of the drug loading. (B) shows various loading curves via different pathways, while intra-maxillary drug releasing type may maintain in a relative continuous and steady level.
Due to slowly and macro-molecularly releasing properties, intra-maxillary pathway may have potential in assistance to ND therapeutic development. Furthermore, the combination therapy within nanotechnology may enhance the efficiency and reduce the relative side effects, including polymer-drug conjugates, nanoemulsions, dendrimers, micelles, liposomes, solid lipid and polymeric nanoparticles [136-138]. Current nanotechnology may even improve the permeability to penetrate the BBB and enhance its concentration in cerebrospinal fluid (CSF) [139]. Personalized drug design according to the inter-individual genetic variability passing through pharmacogenomics may also provide another approach to enhance the drug efficiency [140]. The above developments may be much important especially in long-term administration for ND management to improve the prognosis and promote their life qualities.
Conclusion
Neurodegeneration is currently regarded as a chronic and irreversible process lasting from years to decades. The pathological demyelination, atrophic changes and the related neuronal apoptosis may lead to progressively cognitive impairment and multiple dysfunctions. Due to its specific properties and the related geriatric concerns, the therapeutic design and treatment investigation should be delicately evaluated from research to clinical practice to eliminate the regularly invasive procedures and reduce the side effects. The intra-maxillary molecular releasing provides a long-term continuous and relative painless drug delivery pathway to achieve macromolecular or even cellular delivery into maxilla throughout the dental implant. Since dental implantology has become a mature technique, the combination of the specific nanomedicine and such newly drug delivery pathway may be appropriate for further ND therapeutic applications. However, such device is still immature and multi-disciplinary technology may be involved for further improvement including complementary metal-oxide-semiconductor (CMOS) technology, chemical engineering, pharmacokinetics, therapeutics, medicine, and dentistry. Fortunately, such pathway seems practicable not only theoretically, but also in the medical and dental aspects. As the promotion of life expectance in the beginning of 21st century, ND management may become critical and urgent in geriatric development.
Acknowledgments
Special thanks to Associate Prof. Chih-Cheng Lu of National Taipei University of Technology and his advanced microsystem and device laboratory for the technological support. Valuable assistance from Prof. Jung-Tang Huang of National Taipei University of Technology is also acknowledged. This research received financial support under grants MOST 103-2221-E-027- 017 and MOST 103-2218-E-027-012, Taiwan.
References
- Alzheimer's Association (2008) 2008 Alzheimer’s disease facts and figures. Alzheimers Dement 4(2): 110-133.
- Evans DA, Funkenstein HH, Albert MS, Scherr PA, Cook NR, et al. (1989) Prevalence of Alzheimer’s disease in a community population of older persons:higher than previous reported. J Am Med Assoc 262(18): 2551-2556.
- Hebert LE, Beckett LA, Scherr PA, Evans DA (2001) Annual incidence of Alzheimer’s disease in the United States projected to the years 2000 through 2050. Alzheimer Dis Assoc Disord 15(4): 169-173.
- Crews L, Masliah E (2010) Molecular mechanisms of neurodegeneration in Alzheimer’s disease. Hum Mol Genet 19(R1): R12-20.
- Nelson PT, Braak H, Markesbery WR (2009) Neuropathology and cognitive impairment in Alzheimer disease: a complex but coherent relationship. J Neuropathol Exp Neurol 68(1): 1-14.
- Bertram L, Tanzi RE (2008) Thirty years of Alzheimer’s disease genetics: the implications of systematic meta-analysis. Nat Rev Neurosci 9(10): 768-778.
- Lambert JC, Amouyel P (2011) Genetics of Alzheimer’s disease: new evidences for an old hypothesis? Curr Opin Genet Dev 21(3): 295-301.
- Ittner LM, Gotz J (2011) Amyloid-beta and tau-a toxic pas de deux in Alzheimer’s disease. Nat Rev Neurosci 12(2): 65-72.
- Palop JJ, Mucke L (2010) Amyloid-beta-induced neuronal dysfunction in Alzheimer’s disease: from synapses toward neural networks. Nat Neurosci 13(7): 812-818.
- Alvarez A, Cacabelos R, Sanpedro C, García-Fantini M, Aleixandre M (2007) Serum TNF-alpha levels are increased and correlate negatively with free IGF-1 in Alzheimer’s disease. Neurobiol Aging 28(4): 533-536.
- McGeer EG, McGeer PL (2010) Neuroinflammation in Alzheimer’s disease and mild cognitive impairment: a field in its infancy. J Alzheimers Dis 19(1): 355-361.
- Wyss-Coray T (2006) Inflammation in Alzheimer disease: driving force, bystander or beneficial response? Nat Med 12(9): 1005-1015.
- Mufson EJ, Counts SE, Fahnestock M, Ginsberg SD (2007) Cholinotrophic molecular substrates of mild cognitive impairment in the elderly. Curr Alzheimer Res 4(4): 340-350.
- Schindowski K, Belarbi K, Buee L (2008) Neurotrophic factors in Alzheimer’s disease: role of axonal transport. Genes Brain Behav 7(1): 43-56.
- Kapogiannis D, Mattson MP (2011) Disrupted energy metabolism and neuronal circuit dysfunction in cognitive impairment and Alzheimer’s disease. Lancet Neurol 10(2): 187-198.
- Mosconi L, Berti V, Glodzik L, Pupi A, De Santi S, et al. (2010) Pre-clinical detection of Alzheimer’s disease using FDG-PET, with or without amyloid imaging. J Alzheimers Dis 20(3): 843-854.
- Mosconi L, Pupi A, De Leon MJ (2008) Brain glucose hypometabolism and oxidative stress in preclinical Alzheimer’s disease. Ann N Y Acad Sci 1147: 180-195.
- Smith MA, Zhu X, Tabaton M, Liu G, McKeel DW Jr, et al. (2010) Increased iron and free radical generation in preclinical Alzheimer’s disease and mild cognitive impairment. J Alzheimers Dis 19(1): 363-372.
- Dos Santos GC, Antunes LMG, Dos Santos AC, Bianchi MLP (2009) Coenzyme Q10 and its effects in the treatment of neurodegenerative diseases. Braz J Pharma Sci 45(4): 608-618.
- Bezprozvanny I, Mattson MP (2008) Neuronal calcium mishandling and the pathogenesis of Alzheimer’s disease. Trends Neurosci 31(9): 454-463.
- Bordji K, Becerril-Ortega J, Buisson A (2011) Synapses, NMDA receptor activity and neuronal Aβ production in Alzheimer’s disease. Rev Neurosci 22(3): 285-294.
- Mufson EJ, Counts SE, Perez SE, Ginsberg SD (2008) Cholinergic system during the progression of Alzheimer’s disease: therapeutic implications. Expert Rev Neurother 8(11): 1703-1718.
- Szot P, White SS, Greenup JL, Leverenz JB, Peskind ER, et al. (2006) Compensatory changes in the noradrenergic nervous system in the locus ceruleus and hippocampus of postmortem subjects with Alzheimer’s disease and dementia with Lewy bodies. J Neurosci 26(2): 467-478.
- Hardy JA, Higgins GA (1992) Alzheimer’s disease: the amyloid cascade hypothesis. Science 256(5054): 184-185.
- Lee RK, Wurtman RJ, Cox AJ, Nitsch RM (1995) Amyloid precursor protein processing is stimulated by metabotropic glutamate receptors. Proc Natl Acad Sci USA 92(17): 8083-8087.
- Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, et al. (2003) Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 300(5618): 486-489.
- Kuo YM, Emmerling MR, Vigo-Pelfrey C, Kasunic TC, Kirkpatrick JB, et al. (1996) Water-soluble Abeta (N-40, N-42) oligomers in normal and Alzheimer’s disease brains. J Biol Chem 271(8): 4077-4081.
- Rogers SL, Doody RS, Mohs RC, Friedhoff LT (1998) Donepezil improves cognition and global function in Alzheimer’s disease: a 15-week, doubleblind, placebo-controlled study. Donepezil Study Group. Arch Intern Med 158(9): 1021-1031.
- Blanchard BJ, Konopka G, Russell M, Ingram VM (1997) Mechanism and prevention of neurotoxicity caused by beta-amyloid peptides: relation to Alzheimer’s disease. Brain Res 776(1-2): 40-50.
- Shoji M, Kawarabayashi T, Sato M, Sasaki A, Saido TC, et al. (2000) Agerelated amyloid beta protein accumulation induces cellular death and macrophage activation in transgenic mice. J Pathol 191(1): 93-101.
- Perry G, Rizzuto N, Autilio-Gambetti L, Gambetti P (1985) Paired helical filaments from Alzheimer’s disease patients contain cytoskeletal components. Proc Natl Acad Sci U S A 82(11): 3916-3920.
- Garcia-Sierra F, Ghoshal N, Quinn B, Berry RW, Binder LI (2003) Conformational changes and truncation of tau protein during tangle evolution in Alzheimer’s disease. J Alzheimers Dis 5(2): 65-77.
- Mazanetz MP, Fisher PM (2007) Untangling tau hyperphosphorylation in drug design for neurodegenerative diseases. Nature Review Drug Discovery 6: 464-479.
- Brandt R, Hundelt M, Shahani N (2005) Tau alteration and neuronal degeneration in tauopathies: mechanisms and models. Biochim Biophys Acta 1739(2-3): 331-354.
- Perl DP (2010) Neuropathology of Alzheimer’s disease. Mt Sinai J Med 77(1): 32-42.
- Gendron TF, Petrucelli L (2009) The role of tau in neurodegeneration. Mol Neurodegener 4: 13.
- Iqbal K, Alonso Adel C, Chen S, Chohan MO, El-Akkad E, et al. (2005) Tau pathology in Alzheimer’s disease and other tauopathies. Biochim Biophys Acta 1739(2-3): 198-210.
- Alonso AC, Zaidi T, Grundke-Iqbal I, Iqbal K (1994) Role of abnormally phosphorylated tau in the breakdown of microtubules in Alzheimer’s disease. Proc Natl Acad Sci U S A 91(12): 5562-5566.
- Reddy PH (2011) Abnormal tau, mitochondrial dysfunction, impaired axonal transport mitochondria, and synaptic deprivation in Alzheimer’s disease. Brain Res 1415: 136-148.
- Kosik KS, Orecchio LD, Bakalis S, Neve RL (1989) Developmentally regulated expression of specific tau sequences. Neuron 2(4): 1389-1397.
- Goedert M, Spillantini MG, Jakes R, Rutherford D, Crowther RA (1989) Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron 3(4): 519-526.
- Sennvik K, Boekhoorn K, Lasrado R, Terwel D, Verhaeghe S, et al. (2007) Tau-4R suppresses proliferation and promotes neuronal differentiation in the hippocampus of tau knockin/knockout mice. FASEB J 21(9): 2149-2161.
- Nacharaju P (1999) Accelerated filament formation from tau protein with specific FTDP-17 missense mutations. FEBS letter 447(2-3): 195-199.
- Hutton M (2000) Molecular genetics of chromosome 17 tauopathies. Ann N Y Acad Sci 920: 63-73.
- Hernandez F, Avila J (2007) Tauopathies. Cell Mol Life Sci 64(17): 2219- 2233.
- Churcher I (2006) Tau therapeutic strategies for the treatment of Alzheimer’s disease. Curr Top Med Chem 6(6): 579-595.
- Alvarez XA, Linares C, Masliah E (2012) Combination drug therapy for the treatment of Alzheimer’s disease. European Neurological Review 7(2):92-102.
- Burns A, O’Brien J, Auriacombe S, Ballard C, Broich K, et al. (2006) Clinical practice with anti-dementia drugs: a consensus statement from British Association for Psychopharmacology. J Psychopharmacol 20(6): 732-755.
- Farlow MR, Cummings JL (2007) Effective pharmacologic management of Alzheimer’s disease. Am J Med 120(5): 388-397.
- Hermann N, Li A, Lanctot K (2011) Memantine in dementia: a review of the current evidence. Expert Opin Pharmacother 12(5): 787-800.
- Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH (2010) Mechanisms underlying inflammation in neurodegeneration. Cell 140(6): 918- 934.
- Zach H, Dirkx M, Bloem BR, Helmich RC (2015) The clinical evaluation of Parkinson’s tremor. J Parkinsons Dis 5(3): 471-474.
- Elble R, Bain P, Forjaz MJ, Haubenberger D, Testa C, et al. (2013) Task force report: scales for screening and evaluating tremor: Critique and recommendations. Mov Disord 28(13): 1793-1800.
- De Lau LM, Breteler MM (2006) Epidemiology of Parkinson’s disease. Lancet Neurol 5(6): 525-535.
- Olanow CW, Stern MB, Sethi K (2009) The scientific and clinical basis for the treatment of Parkinson disease. Neurology 72(21 Suppl 4): S1-136.
- Bear MF, Connors BW, Paradiso MA (2007) Neuroscience; exploring the brain. (3rd edtn), Lippincott Williams & Wilkins, Philadelphia. 14: 466-467.
- Dawson TM, Dawson VL (2003) Molecular pathways of neurodegeneration in Parkinson’s disease. Science 302(5646): 819-822.
- Kwakye GF, Paoliello MMB, Mukhopadhyay S, Bowman AB, Aschner M (2015) Manganese-induced Parkinsonism and Parkinson’s disease: shared and distinguishable features. Int J Environ Res Public Health 12(7): 7519-7540.
- Kim YW, Byzova TV (2014) Oxidative stress in angiogenesis and vascular disease. Blood 123(5): 625-631.
- Barbeau A, Roy M, Bernier G, Campanella G, Paris S (1987) Ecogenetics of Parkinson’s disease: prevalence and environmental aspects in rural areas.Can J Neurol Sci 14(1): 36-41.
- Kamel F, Tanner C, Umbach D, Hoppin J, Alavanja M, Blair A, et al. (2007) Pesticide exposure and self-reported Parkinson’s disease in the agricultural health study. Am J Epidermiol 165(4): 364-374.
- Semchuk KM, Love EJ, Lee RG (1992) Parkinson’s disease and exposure to agricultural work and pesticide chemicals. Neurology 42(7): 1328-1335.
- Tanner CM, Ross GW, Jewell SA, Hauser RA, Jankovic J, et al. (2009) Occupation and risk of Parkinsonism: a multicenter case-control study. ArchNeurol 66(9): 1106-1113.
- Parker WD, Boyson SJ, Parks JK (1989) Abnormalities of the electron transport chain in idiopathic Parkinson’s disease. Ann Neurol 26(6): 719-723.
- Schapira AH, Mann VM, Cooper JM, Dexter D, Daniel SE, et al. (1990) Anatomic disease specificity of NADH CoQ1 reductase (complex I) deficiency in Parkinson’s disease. J Neurochem 55(6): 2142-2145.
- Jenner P (2003) Oxidative stress in Parkinson’s disease. Ann Neurol 53(3): 26-38.
- Gary EG, Hsueh-Meei H (2005) Oxidative stress in Alzheimer’s disease. Neurobiol Aging 26: 575-578.
- Butterfield DA (2004) Proteomics: a new approach to investigate oxidative stress in Alzheimer’s disease brain. Brain Res 1000(1-2): 1-7.
- Mckay R, Kittappa R (2008) Will stem cell biology generate new therapies for Parkinson’s disease? Neuron 58(5): 659-661.
- Cardoso F, Seppi K, Mair KJ, Wenning GK, Poewe W (2006) Seminar on Choreas. Lancet Neurol 5(7): 589-602.
- Browne SE, Ferranti RJ, Beal MF (1999) Oxidative stress in Huntington’s disease. Brain Pathol 9(1): 147-163.
- Beal MF (1995) Aging, energy, and oxidative stress in neurodegenerative disease. Ann Neurol 38(3): 357-366.
- Goldberg YP, Telenius H, Hayden MR (1994) The molecular genetics of Huntington’s disease. Curr Opin Neurol 7(4): 325-332.
- Jenkins BG, Rosas HD, Chen YC, Makabe T, Myers R, et al. (1998) 1H NMR spectroscopy studies of Huntington’s disease: correlations with CAG repeat numbers. Neurology 50(5): 1357-1365.
- Kasparova S, Sumbalova Z, Bystricky P, Kucharska J, Liptaj T, et al. (2006) Effect of coenzyme Q10 and vitamine E on brain energy metabolism in the animal model of Huntington’s disease. Neurochem Int 48(2): 93-99.
- Kutzelnigg A, Lucchinetti CF, Stadelmann C, Brück W, Rauschka H, et al. (2005) Cortical demyelination and diffuse white matter injury in multiple sclerosis. Brain 128(Pt 11): 2705-2712.
- Stadelmann C (2011) Multiple sclerosis as a neurodegenerative disease: pathology, mechanisms and therapeutic implications. Curr Opin Neurol 24(3): 224-229.
- Frischer JM, Bramow S, Dal-Bianco A, Lucchinetti CF, Rauschka H, et al. (2009) The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain 132(5): 1175-1189.
- Androdias G, Reynolds R, Chanal M, Ritleng C, Confavreux C, et al. (2010) Meningeal T cells associate with diffuse axonal loss in multiple sclerosis spinal cords. Ann Neurol 68(4): 465-476.
- Magliozzi R, Howell OW, Reeves C, Roncaroli F, Nicholas R, et al. (2010) A gradient of neuronal loss and meningeal inflammation in multiple sclerosis. Ann Neurol 68(4): 477-493.
- Mahad D, Lassmann H, Turnbull D (2008) Review: mitochondria and disease progression in multiple sclerosis. Neuropathol Appl Neurobiol 34(6): 577-589.
- Witte ME, Geurts JJ, de Vries HE, van der Valk P, van Horssen J (2010) Mitochondrial dysfunction: a potential link between neuroinflammation and neurodegeneration? Mitochondrion 10(5): 411-418.
- Anderson JM, Patani R, Reynolds R, Nicholas R, Compston A, et al. (2010) Abnormal tau phosphorylation in primary progressive multiple sclerosis. Acta Neuropathol 119(5): 591-600.
- Hideyama T, Yamashita T, Nishimoto Y, Suzuki T, Kwak S (2010) Novel etiological and therapeutic strategies for neurodiseases: RNA editing enzyme abnormality in sporadic amyotrophic lateral sclerosis. J Pharmacol Sci 113(1): 9-13.
- Rowland LP, Shneider NA (2001) Amyotrophic lateral sclerosis. N Engl J Med 344(22): 1688-1700.
- Sorenson EJ, Stalker AP, Kurland LT, Windebank AJ (2002) Amyotrophic lateral sclerosis in Olmsted County, Minnesota, 1925-1998. Neurology 59(2): 280-282.
- Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, et al. (1993) Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362(6415): 59-62.
- Bogdanov M, Brown RH, Matson W, Smart R, Hayden D, et al. (2000) Increased oxidative damage to DNA in ALS patients. Free Radic Biol Med 29(7): 652-658.
- Beal MF, Ferrante RJ, Browne SE, Matthews RT, Kowall NW, et al. (1997) Increased 3-nitrotyrosine in both sporadic and familial amyotrophic lateral sclerosis. Ann Neurol 42(4): 644-654.
- Ferrante RJ, Browne SE, Shinobu LA, Bowling AC, Baik MJ, et al. (1997) Evidence of increased oxidative damage in both sporadic and familial amyotrophic lateral sclerosis. J Neurochem 69(5): 2064-2074.
- Murata T, Ohtsuka C, Terayama Y (2008) Increased mitochondrial oxidative damage in patients with sporadic amyotrophic lateral sclerosis. J Neurol Sci 267(1-2): 66-69.
- Enciu AM, Nicolescu MI, Manole CG, Muresanu DF, Popescu LM, et al. (2011) Neuroregeneration in neurodegenerative disorders. BMC Neurol 11: 75-81.
- Dantuma E, Merchant S, Sugaya K (2010) Stem cells for the treatment of neurodegenerative diseases. Stem Cell Research & Therapy 1: 37-43.
- Andersen JK (2004) Oxidative stress in neurodegeneration: cause or consequence? Nat Med 10: 18-25.
- Somayajulu M, Mccarthy S, Hung M, Sikorska M, Borowy-Borowski H, et al. (2005) Role of mitochondria in neuronal cell death induced by oxidative stress; neuroprotection by Coenzyme Q10. Neurobiol Dis 18(3): 618-627.
- Mellors A, Tappell AL (1966) The inhibition of mitochondrial peroxidation by ubiquinone and ubquinol. J Biol Chem 241(19): 4353-4356.
- Takayanagi R, Takeshige K, Minakami S (1980) NADH- and NADPHdependent lipid peroxidation in bovine heart submitochondrial particles. Dependence on the rate of electron flow in the respiratory chain and an antioxidant role of ubiquinol. Biochem J 192(3): 853-860.
- Shults CW, Oakes D, Kieburtz K, Beal MF, Haas R, et al. (2002) Effects of Coenzyme Q10 in early Parkinson disease: evidence of slowing of the functional decline. Arch Neurol 59(10): 1541-1550.
- Muller T, Buttner T, Gholipour AF, Kuhn W (2003) Coenzyme Q10 supplementation provides mild symptomatic benefit in patients with Parkinson’s disease. Neurosci Lett 341(3): 201-204.
- Isharat T, Khan MB, Hoda MN, Yousuf S, Ahmad M, et al. (2006) Coenzyme Q10 modulates cognitive impairment against intracerebroventricular injection of streptozotocin in rats. Behav Brain Res 171(1): 9-16.
- Koroshetz WJ, Jenkins BG, Rosen BR, Beal MF (1997) Energy metabolism defects in Huntington’s disease and effects of Coenzyme Q10. Ann Neurol 41(2): 160-165.
- Galpern WR, Cudkowicz ME (2007) Coenzyme Q treatment of neurodegenerative diseases of aging. Mitochondrion 7: 146-153.
- Southwell AL, Patterson PH (2010) Antibody therapy in neurodegenerative disease. Rev Neurosci 21(4): 273-287.
- Lemere CA, Masliah E (2010) Can Alzheimer’s disease be prevented by amyloid-beta immunotherapy? Nature Rev Neurol 6(2): 108-119.
- Westwood M, Lawson ADG (2015) Opportunities for conformation-selective antibodies in amyloid-related diseases. Antibodies 4(3): 170-196.
- Lemere CA (2013) Immunotherapy for Alzheimer’s disease: hoops and hurdles. Mol Neurodegener 8: 36-42.
- Lannfelt L, Moller C, Basun H, Osswald G, Sehlin D, et al. (2014) Perspectives on future Alzheimer’s therapies: amyloid-β protofibrils- a new target for immunotherapy with BAN2401 in Alzheimer’s disease. Alzheimers Res Ther 6(2): 16-24.
- Meli G, Lecci A, Manca A, Krako N, Albertini V, et al. (2014) Conformational targeting of intracellular Aβ oligomers demonstrates their pathological oligomerization inside the endoplasmic reticulum. Nat Commun 5.
- Dodel R, Hampel H, Depboylu C, Lin S, Gao F, et al. (2002) Human antibodies against amyloid beta peptide: a potential treatment for Alzheimer’s disease. Ann Neurol 52(2): 253-256.
- Fillit H, Hess G, Hill J, Bonnet P, Toso C (2009) IV immunoglobulin is associated with a reduced risk of Alzheimer’s disease and related disorders. Neurology 73(3): 180-185.
- Relkin NR, Szabo P, Adamiak B, Monthe C, Lent RW, et al. (2009) 18-month study of intravenous immunoglobulin for treatment of mild Alzheimer’s disease. Neurobiol Aging 30(11): 1728-1736.
- Asuni AA, Boutajangout A, Quartermain D, Sigurdsson EM (2007) Immunotherapy targeting pathological tau conformers in a tangle mouse model reduces brain pathology with associated functional improvements. J Neurosci 27(34): 9115-9129.
- Miller TW, Shirley TL, Wolfgang WJ, Kang X, Messer A (2003) DNA vaccination against mutant Huntington ameliorates the HDR6/2 diabetic phenotype. Mol Ther 7(5 Pt 1): 572-579.
- Labbadia J, Morimoto RI (2013) Huntington’s disease: underlying molecular mechanisms and emerging concepts. Trends Biochem Sci 38(8): 378- 385.
- Lashuel HA, Overk CR, Oueslati A, Masliah E (2013) The many faces of α-synuclein: from structure and toxicity to therapeutic target. Nat Rev Neurosci 14: 38-48.
- Tuszynski MH, Thal L, Pay M, Salmon DP, Bakay R, et al. (2005) A phase 1 clinical trial of nerve growth factor gene therapy for Alzheimer’s disease. Nat Med 11(5): 551-555.
- Bjorklund LM, Sanchez-Pernaute R, Chung S, Andersson T, Chen IY, et al. (2002) Embryonic stem cells develop into functional dopaminergic neurons after transplantation in a Parkinson rat model. Proc Natl Acad Sci USA 99(4): 2344-2349.
- Wang Q, Matsumoto Y, Shindo T, Miyake K, Shindo A, et al. (2006) Neural stem cells transplantation in cortex in a mouse model of Alzheimer’s diseases. J Med Invest 53(1-2): 61-69.
- Sadan O, Shemesh N, Cohen Y, Melamed E, Offen D (2009) Adult neurotrophic factor-secreting stem cells: a potential novel therapy for neurodegenerative diseases. Isr Med Assoc J 11(4): 201-204.
- Reynolds BA, Weiss S (1992) Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science 255(5052): 1707-1710.
- McKay R (1997) Stem cells in the central nervous system. Science 276(5309): 66-71.
- Shihabuddin LS, Palmer TD, Gage FH (1999) The search for neural progenitor cells: prospects for the therapy of neurodegenerative disease. Mol Med Today 5(11): 474-480.
- Van Strien ME, Sluijs JA, Reynolds BA, Steindler DA, Aronica E, et al. (2014) Isolation of neural progenitor cells from the human adult subventricular zone based on expression of the cell surface marker CD271. Stem Cells Transl Med 3(4): 470-480.
- Ma DK, Bonaguidi MA, Ming GL, Song H (2009) Adult neural stem cells in the mammalian central nervous system. Cell Res 19(6): 672-682.
- Zhao C, Deng W, Gage FH (2008) Mechanisms and functional implications of adult neurogenesis. Cell 132(4): 645-660.
- Mimeault M, Hauke R, Batra SK (2007) Stem cells: a revolution in therapeutics- recent advances in the stem cell biology and their therapeutic applications in regenerative medicine and cancer therapies. Clin Pharmacol Ther 82(3): 252-264.
- Gao YH, Li XC, Zheng D, Guan WJ, Ma YH (2015) Isolation of a pluripotent neural stem cell from the embryonic bovine brain. Int J Mol Sci 16(3):5990-5999.
- Yamanaka S (2009) A fresh look at iPS cells. Cell 137(1): 13-17. [129]. Xiao J, Yang RB, Biswas S, Qin X, Zhang M, et al. (2015) Mesenchymal stem cells and induced pluripotent stem cells as therapies for multiple sclerosis. Int J Mol Sci 16(5): 9283-9302.
- Mattson MP (2000) Neuroprotective signaling and the aging brain: take away my food and let me run. Brain Res 886(1-2): 47-53.
- Cole GM, Frautschy SA (2007) The role of insulin and neurotrophic factor signaling in brain aging and Alzheimer’s disease. Exp Gerontol 42(1-2): 10-21.
- Peterson AL, Nutt JG (2008) Treatment of Parkinson’s disease with trophic factors. Neurotherapeutics 5(2): 270-280.
- Yasuyuki S, Masaki K, Junichiro T, Tomoko A, Maho M, et al. (2009) Analysis of 472 Brånemark system TiUnite implants: A retrospective study. Kobe J Med Sci 55(3): 73-81.
- Lu CC, Li YJ, Tsai WL, Chang CK (2015) An implantable and painless drug delivery device for long-term operation. Taiwan Patent I488620.
- Li YJ, Lu CC (2015) A novel scheme and evaluations on a long-term and continuous biosensor platform integrated with a dental implant fixture and its prosthetic abutment. Sensors 15(10): 24961-24976.
- Estanqueiro M, Amaral MH, Sousa Lobo JM (2015) New trends in pharmaceutical nanotechnology. Curr Pharm Des 21(36): 5169-5171.
- Ho LC, Hsu CH, Ou CM (2015) Unibody core-shell smart polymer as a theranostic nanoparticle for drug delivery and MR imaging. Biomaterials37: 436-446.
- Gu X, Ding J, Zhang Z, Li Q, Zhuang X, Chen X (2015) Polymeric nanocarriers for drug delivery in osteosarcoma treatment. Curr Pharm Res 21(36): 5187-5197.
- Agrawal U, Chashoo G, Sharma PR, Kumar A, Saxena AK, et al. (2015) Tailored polymer-lipid hybrid nanoparticles for the delivery of drug conjugate: dual strategy for brain targeting. Colloids Surf B Biointerfaces 126: 414-425.
- Thompson DK (2016) From research to clinical application: challenges in regulating companion biomarker tests for “personalized” drugs. J Pharm Anal Insights 1(1): 1-2.





