Toxic Compounds Analysis With High Performance Liquid Chromatography Detected By Electro Chemical Detector (Ecd)
Hideharu Shintani
1 Faculty of Science and Engineering, Chuo University, 1-13-27, Kasuga, Bunkyo, 112-8551, Tokyo, Japan
*Corresponding Author
Hideharu Shintani,
Faculty of Science and Engineering,
Chuo University, 1-13-27, Kasuga,
Bunkyo, 112-8551, Tokyo, Japan.
Tele/Fax: +81425922336.
E-mail: shintani@mail.hinocatv.ne.jp
Article Type: Review Article
Received: September 17, 2014; Accepted: October 01, 2014; Published: October 02, 2014
Citation: Hideharu Shintani (2014) Toxic Compounds Analysis With High Performance Liquid Chromatography Detected By Electro ChemicalDetector (Ecd). Int J Clin Pharmacol Toxicol. 3(3), 121-127. doi: dx.doi.org/10.19070/2167-910X-1400022
Copyright: Hideharu Shintani© 2014. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
Abstract
The principal area of application of high performance liquid chromatography-electrochemical detector (HPLC-ECD) has been in the analysis of naturally-occurring analytes, such as catecholamines, and pharmaceuticals in biological samples, HPLC-ECD has also applied to the analysis of pesticides and other analytes of interest to the toxicologist. In this paper, toxic area is described. In these, ammatoxins, aromatic amine, nitro-compounds, algal toxins, fungal toxins, pesticides, veterinary drug and food residues will be concretely described.
2.Introduction
3.Toxic compound analysis by ECD
4.Alpha Ammatoxins In Urine
5.Aromatic Amines and Nitro-compounds
6.Algal and FungaI Toxins
7.Pesticides and Related Compounds
8.Veterinary Drug and other Food Residues
9.Conclusion
10.References
Keywords
Electrochemical detection, ECD, HPLC, Toxic compound, Toxicology.
Introduction
Toxic compound analysis produced by sterilization and electrochemical detection (ECD) was omitted in this paper because that subject was discussed in the editorial. They are mostly from the author [1].
ECD is used for the sensitive and selective detection and measurement of electro-active analytes in many areas of analytical chemistry and biochemistry. These applications range from electrode sensor devices via flow injection analyses (FIA) to direct measurements of neurochemicals in brain tissue using in vitro cyclic voltammetry. In separation science, ECD is used to detect and measure responsive analytes in flowing streams following analysis by high-performance liquid chromatography (HPLC) or capillary electrophoresis (CE). The use of ECD with HPLC is by far and away the most important application. However, its popularity should be compared to HPLC in combination with fluorescence detection and with MS detection and the most used methodology is HPLC combined with UV/visible detection.
Unlike UV or fluorescence detectors, ECD does not exploit a physical property of an analyte, but an induced chemical change that results from an electrochemical reaction. ECD differs from other reactors used in HPLC in that requirement of ionic reagent in the mobile phase for electronic detection or reaction devices are normally required to effect the chemical change in the analyte such as keto-enol or amino-imino tautomerism). In addition, the reaction kinetics are usually fast leading to minimal extra-column effects.
The principles of ECD when specifically applied to HPLC and/or CE have been already described.
With UV detectors, selectivity is adjusted by varying the detection wavelength, lower wavelengths often giving enhanced sensitivity and a response from a wider range of analytes, but selectivity will be lost. A modest degree of selectivity is achieved by using UV detection in the aromatic region (240-270 nm) and traditionally 254 nm for compound with benzene ring has proved popular. However at lower wavelengths (200-210 nm) the absorption of the eluent of other eluent constituents or of oxygen become limiting, therefore degassing equipment is required. Relatively few compounds show useful absorption at wavelengths higher than 340 nm (the lower limit of the deuterium lamp emission). Generally, responses are usually independent of eluent conditions. In ECD detection both sensitivity and selectivity are adjusted by varying the potential maintained between the working and reference electrodes, higher potentials, up to a local maximum, giving increased sensitivity with increasing applied potential. However, higher potentials usually induce a response from more compounds and therefore compromise selectivity. In oxidative mode, oxidation of eluent constituents becomes limiting at higher applied potentials, whilst in reductive mode, interference from dissolved oxygen can prove difficult to exclude. The response at the electrode is also very dependent on the eluent composition especially its pH. Thus, as in all analytical methods it is the signal-to-noise (S/N) ratio that is important and the detection conditions eventually adopted for a separation are a compromise between the electrochemical response of the analyte, the optimum eluent for both detection and elution, and interference from the sample matrix or from noise or drift from electronic or other sources.
In HPLC-ECD the column eluate flows over the surface of an inert electrode maintained at an appropriate positive or negative potential relative to a reference electrode. At the electrode surface analytes possessing electroactive functional groups undergo oxidation or reduction. The electrons released (or donated) travel via the electrode and the change in current can be measured and related to the concentration of the analyte. Modem electronics allow the applied potential to be held within very tight limits while at the same time measuring and amplifying the very small currents created. Hence these detectors can be very sensitive. A crude comparison of the sensitivity and applicability of the most common HPLC detectors towards favored analytes under similar analytical conditions is given. Both ECD and fluorescence detectors can be at least 50-100 times more sensitive towards responsive compounds than a standard UV detector and are much more selective. Unfortunately, with time ECD reaction products tend to accumulate at the electrode surface leading to loss of activity and hence loss of detector response. This is the major reason ECD remains a relatively specialized field.
Toxic compound analysis by ECD
HPLC is widely used in analytical toxicology. UV/Visible absorption (including photo diode-array and scanning instruments) and fluorescence detection remain of paramount importance, with pre- or post-column derivatization sometimes being used to enhance sensitivity and/or selectivity. Modem UV detectors are in the main considerable improvement on their predecessors. HPLC-MS, HPLC-MS-MS or HPLC-TOFMS are being used increasingly in quantitative work, although the capital costs involved remain relatively high. Nevertheless, ECD still finds a role in certain applications and there is a considerable body of literature associated with this topic. ECD requires more care and thought in routine use than spectrophotometric detectors, principally because of the problems of electrode deactivation. on the other hand, running costs can be minimal and good sensitivity/selectivity can be attained with a number of analytes.
The aim of this article is to give information to aid the use of HPLC-ECD in the analysis of drugs and poisons in biological and related specimens.The available information (column,eluent, detection potential, extraction procedure, internal standard,sensitivity, etc.) is presented. Sufficient information is given for the reader to decide whether a particular approach is worth pursuing. Chemical names or structural formulae are given to aid identification of electroactive moieties. The use of alternative techniques,including CE-ECD is emphasised as appropriate. Additional topics, such as analyte stability, are also discussed where relevant.Note that unambiguous details of the working and reference electrode combinations used in a particular application are not always given in published work-in such cases an informed guess as to the ECD conditions actually used has had to be made.
Alpha Ammatoxins In Urine
The study was to develop and validate a liquid chromatographic method with electrochemical detection to measure α-amanitin concentrations in urine after sample pretreatment with double mechanism (reversed phase/cation exchange) solid-phase extraction cartridges. The urine samples (10 mL)were purified and concentrated to l mL with elimination of matrix contaminants. The extracts were then separated by isocratic reversed-phase chromatography using a C18 column (4.6mmx25cm) with a mobile phase composed of 0.005 M phosphate buffer (pH 7.2) and acetonitrile (9/1). Coulometric detection was performed by applying an oxidation potential of +500 mV to a porous graphite electrode in a low-volume analytical cell. The limit of quantitation was 10 ng/ ml with a signal to-noise ratio of 3 (S/N=3). The linearity studied on spiked urine was satisfactory (r=O.9966)from 10 ng/mL to 200 ng/mL. The average extraction recovery of α-amanitin was 78%, determined using spiked urine samples ranging from 10- 300 ng/mL. The intra-assay precision was checked at 10,50 and 100 ng/mL levels(n=10) in spiked urine samples, with resulting coefficients of variation of 3.6%,2%and l.5%,respectively[2]. The chemical structure is shown in Figure 1.
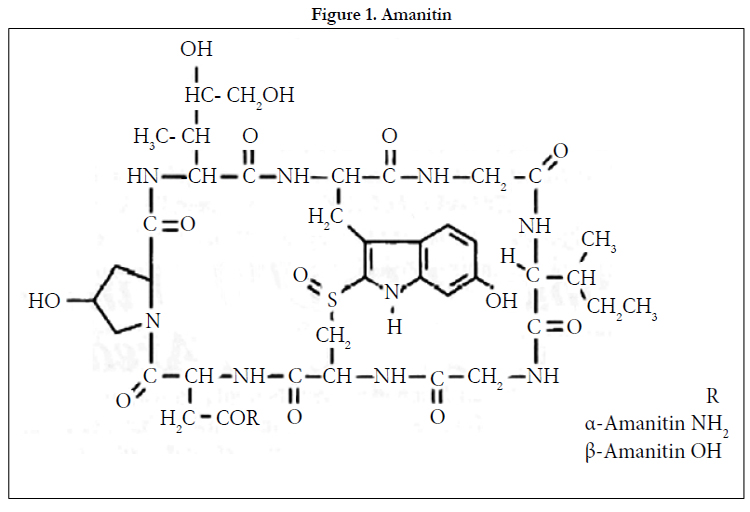
Figure 1. Amanitin.
Aromatic Amines and Nitro-compounds
Felice et al [3] used a glassy carbon electrode (GCE, +0.7 to +0.9 V vs Ag/AgCI) in the analysis of polycyclic aromatic amines, such as 2-aminonaphthalene, 4-aminobiphenyl,and 2-aminoanthracene,in rodent skin samples after topical application of these compounds. The HPLC system used consisted of a C18-modined silica column with acetonitrile-aqueous citrate/perchlorate buffer (7/3 or thereabouts) as eluent-the buffer composition and the proportion of acetonitrile were varied in different experiments. A limit of detection (LOD) of 0.l pmol on column could be expected.
Heterocyclic aromatic amines, such as 2-amino- 3-methylimidazo(4,5-f) quinoline,arise from amino acids,proteins, etc during cooking. Billedeau et al[4] used a short-chain alkyl-modified (SynChropak SCD-100) analytical column and ECD (GCE, +0.6-0.9 V vs Ag/AgCl) in the analysis of these compounds. The eluent was acetonitrile-aqueous ammonium acetate(50 mmoI L-1, pH 5.5-6.5)(1/3, v/v).No results of sample analyses were reported.
Nitrated polycyclic aromatic hydrocarbons, such as 1-nitropyrene, 1-nitrosopyrene and l,3-dinitropyrene, have been measured in car exhaust deposits by HPLC with chemiluminescence detection after on-line ECD reduction (PGEs, -1.6V vs Pd)[5]. LODs of fmol on-column were claimed.
Algal and FungaI Toxins
The trichothecene mycotoxins deoxynivalenol (vomitoxin), nivalenol and fusarenon-X (Figure 2) have been measured in wheat extracts using HPLC with post-column photolysis (Hg vapor lamp, 2.3-3 min residence time) and ECD (paralle1 GCE, +1.1. V and +0.85 V vs Ag/AgCl)[6]. A C18-modified silica analytical column was used with methanol-aqueous sodium chloride (50 mmol L-1) (15 /85) as eluent. LODs were 1-2 ng on-column.
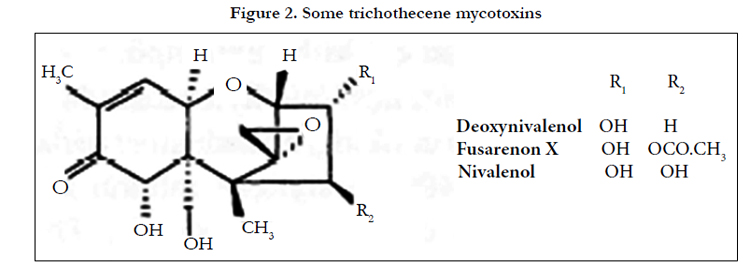
Figure 2. Some trichothecene mycotoxins.
The mycotoxin agaritine (β-N-(γ-L (+) glutamy1)-4-hydroxymethyl phenylhydrazine, (Figure 3 upper) been measured in cultivated mushrooms using a C18-modified silica column with methanol-aqueous potassium hydrogen phosphate (50 mmol L-1) (2.5/97.5, v/v) as eluent and ECD (PGEs,E1:+0.6 vs Pd) [7]. Sample preparation (fresh produce) was by homogenization and methanol extraction. No internal standard (IS) was used. The LOD was 5 mg kg-1 dry weight.
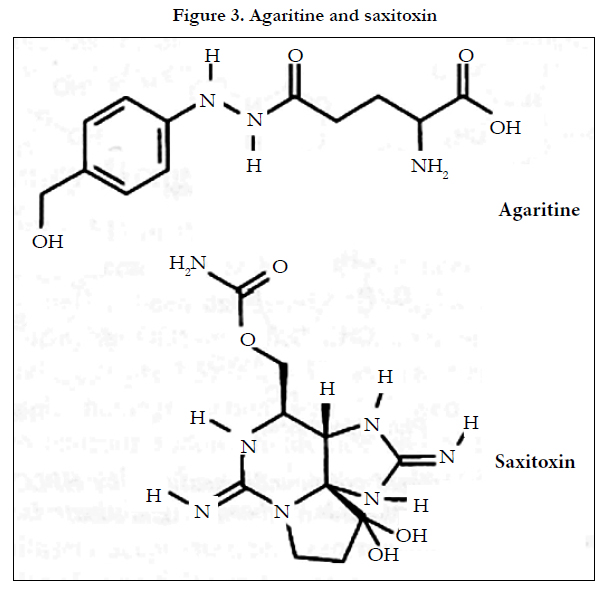
Figure 3. Agaritine and saxitoxin.
Saxitoxin (Figure 3 lower) and several other algal toxins that are known to sometimes contaminate shellfish have been measured by fluorescence (excitation 330 nm; emission 395 nm) and mass spectrometric detection after electrochemical oxidation in an explicit semantic analysis (ESA) 5020 guard cell (PGE +1.05 V vs Pd)[8]. Chromatographic analysis was achieved using anionexchange and cation-exchange columns connected in series with gradient elution (20-450 mmol L-1 aqueous ammonium acetate, pH6.9). LODs (S/N=3) for saxitoxin were claimed to be 0.03 ng (fluorescence) and 0.5 ng (Mass-spectrometry, MS).The method was applied to the measurement of the toxins in mussels.
Pesticides and Related Compounds
Chlorophenols, such as 2,4-dichlorophenol, occur as impurities in chlorophenoxy herbicide formulations at concentrations of about 1%(w/w) of the herbicides. Akerblom and Lindgren[9]used seria1 UV (280 nm) and ECD (carbon paste electrode, +1.05 V vs Ag/AgCl) for the herbicides and chlorophenols, respectively. The analytical column was C18-modified silica and the eluent was methanol-aqueous acetic acid (0.1 mol L-1) (9/11 to 11/9 depending on the formulation under analysis). This approach could prove useful for the analysis of these compounds in biological specimens.
The insecticide carbaryl (1-naphthyl-N-methylcarbamate, Figure 4) has been measured on filter paper exposed during aerial spraying using a C18-modified silica column and ECD (GCE, +0.75 V vs Ag/AgCl) after pre-column alkaline hydrolysis to l-naphthol [10]. The eluent was acetonitrile acetic acid-aqueous potassium chloride (0.5 mo1 L-1) (50/1/49). A 2-naphthol was IS. Postcolumn hydrolysis with sodium hydroxide of carbaryl and some other N-methyl-carbamate pesticides has also been employed in measuring crop residues of these compounds[11]. The analytical column used was octyl(C8)-modified silica. Elution was by means of an acetonitrile-water gradient and ECD was at PGEs (E1:+0.20 V, E2:+0.60 V vs Pd).
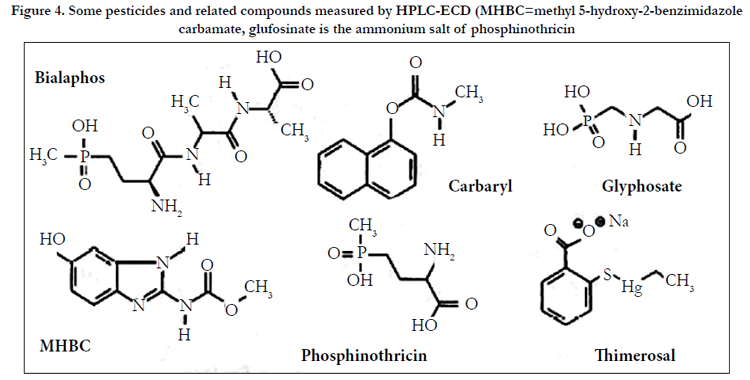
Figure 4. Some pesticides and related compounds measured by HPLC-ECD (MHBC=methyl 5-hydroxy-2-benzimidazole carbamate, glufosinate is the ammonium salt of phosphinothricin.
Methyl 5-hydroxy-2-benzimidazole carbamate (MHBC, Figure 4), a urinary metabolite of the pesticides carbendazium, benomyl and thiophanate-methyl, has been measured using a C18-modified silica analytical column with methanol-aqueous ammonium acetate (approximately 60 mmoI L-1, pH 8) (27/73, v/v) as eluent and ECD (PGE, +0.22V vs Pd)[12]. Sample preparation was by a complex procedure involving SPE (SCX-modified silica) of hydrolyzed specimens. SPE and SCX are solid phase extraction and strong cation exchange. No IS was used. The LOD was 5 μgL-1(l mL sample).
The HPLC-ECD of the herbicides glyphosate, glufosinate (the ammonium salt of phosphinothricin), and bialaphos (Figure 4) using integrated pulsed amperometric detection (IPAD) has been described[13]. The analytes were analyzed using an IonPacR AS15 anion-exchange column with gradient elution (purified water/ aqueous sodium hydroxide (0.2 mol L-1)). Detection was by adsorption onto a gold working electrode at -0.3 V and oxidation at +0.23 V vs Ag/AgCl. The electrode surface was cleaned and reactivated by applying potentials of –0.2 V and+0.63 V, before being returned to -0.3V. LODs of 50, 20 and 65 μgL-1 (S/N=3) were claimed for glyphosate, glufosinate and bialaphos, respectively. The method was applied to spiked human urine and plasma and to urine from a mouse dosed with glyphosate (750 mg kg-1).
The antiseptic thimerosal (sodium ethylmercury thiosalicylate, Figure 4) and its major degradation products thiosalicylic acid and 2,2’-dithiodibenzoic acid have been studied in pharmaceutical preparations using HPLC-ECD (C18-modified silica or anionexchange resin) [14]. Glassy carbon or carbon paste electrodes in wall-jet or thin-layer configuration were used (+0.9V vs Ag/AgCl (+1.2 V for 2,2'-dithiodibenzoic acid)) in addition to PGEs (+0.8 V vs Pd, all analytes). LODs (PGE) ranged from 2 to 20 μgL-1. A similar approach was adopted by Kang and Kim[15] to measure methylparaben and propylparaben (methyl and propyl 4-hydroxybenzoate) and thimerosal, again in pharmaceutical preparations. Several C18-based columns and eluents of pH 2.5-6.0 were evaluated. Detection was at a GCE (+1.25 V vs Ag/AgCl).
Veterinary Drug and other Food Residues
The use of HPLC-ECD in the analysis of the anabolic steroids and metabolites, diethylstilbestrol, taleranol, zearalenol, zearalenone and zeranol (Figure 5), in mammalian tissue has been discussed [16]. Diethylstilbestrol has been measured in animal tissue using a C18-modified silica column with methanol-aqueous phosphate buffer (50 mmol L-1, pH 3,5) (67/33. v/v) as eluent and ECD (GCE, +0.9 V vs Ag/AgCl) [17]. Sample homogenisation was followed by liquid-liquid extraction (LLE) into methyl tertiary butyl ether (MTBE), back-extraction into aqueous sodium hydroxide and solid phase extraction (SPE, C18-modified silica). The LOD was approximately 0.5 μgkg-1 wet weight (10 g sample).
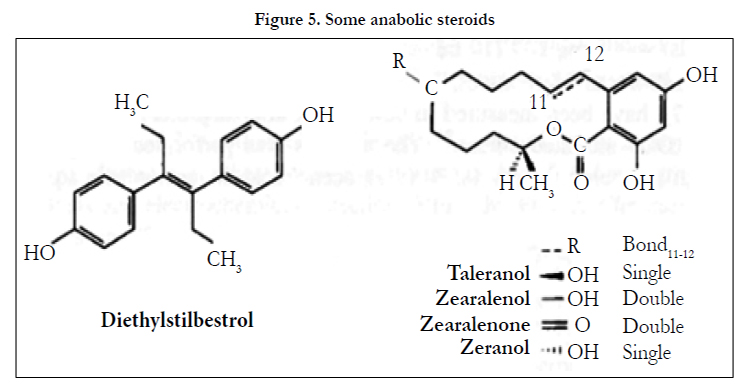
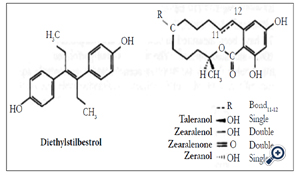
Figure 5. Some anabolic steroids.
The tranquillisers acepromazine, azaperone, chlorpromazine, haloperidol, propionylpromazine and xylazine (Figure 6), the azaperone metabolite/tautomer azaperol and the β-adrenoreceptor blocker carazolol have been measured in animal tissue using a methyl- modified silica analytical column with acetonitrile-aqueous ammonium acetate (1.54 g L-1) (1/1, v/v) as eluent with ECD(PGEs, E1: +0.4 V, E2: +0.7 V vs Pd)[18]. Homogenized tissue was extracted using SPE (C18-modified silica). LODs were 2μgkg-l wet weight (5 g samples).
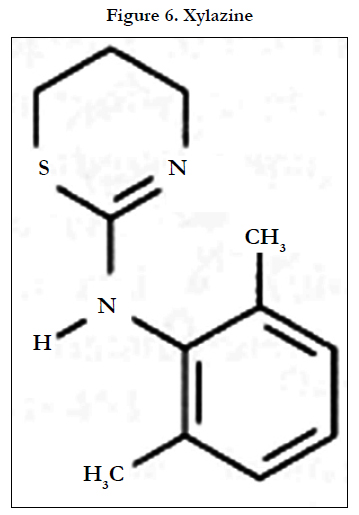
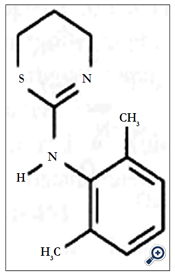
Figure 6. Xylazine.
Gossypol and gossypolone (gossypol in which the hydoxyl moieties on the two inner aromatic rings are replaced by quinones) enantiomers have been measured in fish using HPLC with ECD and UV detection [19]. The analytes were derivatized prior to analysis on C18-modified silica column with acetonitrile-water (8/1.v/v) containing aqueous potassium dihydrogen orthophosphate (2 mmoI L-1), final pH 3.0 adjusted with orthophosphoric acid, as eluent. Blood, seminal plasma and kidney homogenates were extracted with dimethylformamide-(R(-)-2-amino-1-propanol)-acetic acid(44/1/5, v/v/v) and heated (90-95°C, 30 min).Detection was at a GCE (+0.85V vs Ag/AgCl). LODs (S/N=3) using ECD were 2.5 and 50μgL-l for gossypol and gossypolone, respectively, some16 and 6 times lower than those obtained by UV. Sample sizes and extraction volumes were not given.
A number of penicillin antibiotics, including amoxicillin, ampicillin, cloxacillin and penicillin G (benzylpenicillin) have been measured in milk using a C18-modified silica column with gradient elution (acetonitrile-methano1-aqueous acetate buffer (20 mmoI L-1) and PAD (pulsed amperometric detection, serial dua1 Au electrodes, Ag/AgCI reference)[20]. Sample preparation was by preconcentration of the analytes on the analytical column after protein precipitation with acetonitrile and fat extraction using dichloromethane-hexane (1/1. v/v). LODs were of the order of 0.2μmo1 L-1. A similar approach was adopted by Dasenbrock and LaCourse[21] for the measurement of cephapirin and ampicillin in milk. Isocratic elution with acetonitrile-aqueous acetate buffer (0.5 moI L-1, pH 3.75)-water(1/5/19, v/v/v) and an octyl (C8)-modified silica column was used. Detection was by integrated pulsed amperometric detection (IPAD) at a gold electrode; four sweeps between +0.05 to +1.15 V vs Ag/AgCl were integrated. The LOD (both compounds)was 5 mg L-1 (10 mL sample).
Three nitrofuran derivatives, nitrofurantoin, furazolidone and furaltadone (Figure 7), have been measured in cow's milk after deproteinisation with trichloro acetic acid (TCA) and SPE (C18- modifiedsilica)[22].The analysis was performed using a C18- modified silica column with 0.5%(v/v) acetic acid in acetonitrileaqueous sodium perchlorate (0.l moI L-1) (7/18, v/v). Oxygen was removed by bubbling with nitrogen. Three electrode PGE detection was described (ESA 5021 conditioning cell and ESA 5011 high sensitivity analytical cell), but only one working potentia1, -0.6V vs Pd, was quoted. LODs of 4-6 μgL-l were claimed (50 mL sample). Interestingly, this same group used a very similar approach to measure nitro pesticides in river water[23]. The conditioning cell was operated at -0.6V vs Pd to contribute to the elimination of oxygen in the mobile phase. The analytical cell was operated in the redox mode (E1: -1.3 V E2: +0.8V vs Pd), the signals from both electrodes being used to quantify the compounds.
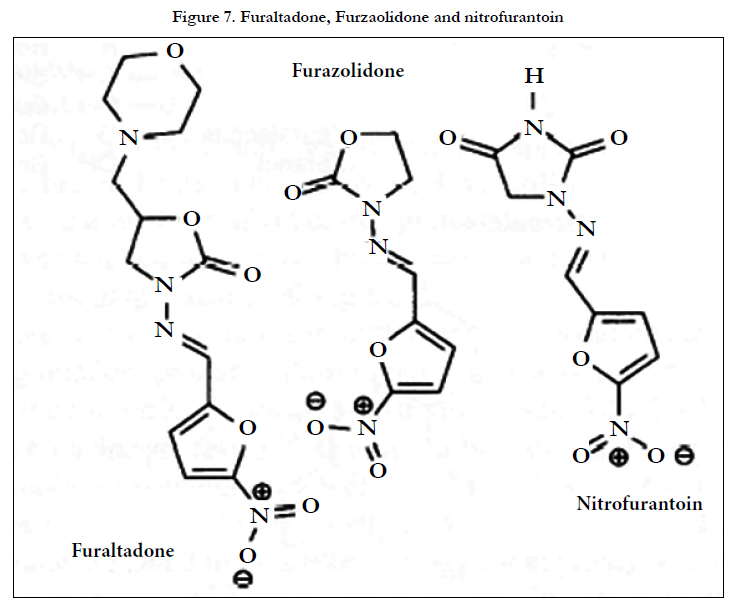
Figure 7. Furaltadone, Furzaolidone and nitrofurantoin.
Conclusion
The purpose of this article is to discuss applications of HPLCECD in the analysis of toxic compounds and poisons in biologicalb specimens,usually plasma or urine. Analytes are mainly toxic compounds and poisons in nature. Chemical structures are given for most analytes and IS,as are ECD conditions, column,eluent, sample size and limit of detection (LOD) and low limit of quantitation (LLOQ).Brief details of sample preparation procedures were also included in many instances.The ECD for toxic compounds or poisons with the aim of increasing sensitivity/selectivity was specially given attention. The use of serial GCEs is also under investigation. However,use of ECD in conjunction with mini-bore (2mm i.d.) or narrower columns provides a means of enhancing sensitivity with existing equipment.
The use of ECD either in the form of pyrolytic graphite electrode arrays or technically simpler systems in the qualitative analysis of toxic compounds or poisons in urine, possibly as an adjunct to photo diode array or scanning UV detection, is a further topic. An additional area where developments might be expected is in the application of post-column photolysis to generate electroactive species.
References
- H.Shintani (3024) Editorial, Toxic Compounds Analysis by High Performance Liquid Chromatography, International J. Clin. Pharm. Toxicol., in submission.
- C.Defendenti, E.Bonacina, M.Mauroni, L. Gelosa (1998) Validation of a high performance liquid chromatographic method for alpha-amanitin determination in urine. Forensic Sci. Int. 92:59-68.
- L.J. Felice, R.E. Schirmer, D.L. Springer, C.V. Veverka (1986) Determination of polycyclic aromatic amines in skin by liquid chromatography withelectrochemical detection. J. Chromatogr. 354:442-448.
- S.M. Billedeau, M.S. Bryant, C. L. Holder (1991) Analysis of heterocyclic amines using reversed-phase high performance liquid chromatography with electrochemical detection. LC-GC Intl. 4:38-41.
- N. Imaizumi, K.Hayakawa, Y. Suzuki, M. Miyazaki (1990) Determination of nitrated pyrenes and their derivatives by high performance liquid chromatography with chemiluminescence detection after online electrochemical reduction. Bioomed. Chromatogr. 4:108-112.
- W.L. Childress, I.S. Krull, C.M. Selavka. (1990) Determination of deoxynivalenol (DON, vomitoxin) in wheat by high-performance liquid chromatography with photolysis and electrochemical detection (HPLC-hv-EC). J. Chromatogr. Sci. 28:76-82.
- M. Sharman, A.L. Patey and J.Gilbert (1990) A survey of the occurrence of agaritine in U.K. cultivated mushrooms and processed mushroom products, Food Addit. Contam. 7:649-656.
- E. Jaime, C. Hummert, P.Hess, B. Luckas. (2001) Determination of paralytic shellfish poisoning toxins by high-performance ion-exchange chromatography. J. Chromatogr. 929:43-49.
- M. Akerblom, B.Lindgren (1983) Simultaneous determination of active ingredients and chlorophenol impurities in phenoxy acid herbicide formulations by high-performance liquid chromatography with ultra-violet and electro-chemical detection. J. Chromatogr. 258:302-306.
- S. Kawai, K.Goto, K. Kano, T. Kubota (1988) Determination of carbaryl by high-performance liquid chromatography with electrochemical detection. J. Chromatogr. 442:451-454.
- R.T.Krause. (1988) High-performance liquid chromatographic determination of aryl-N-methylcarbamate residues using post-column hydrolysis electrochemical detection. J.Chromatogr. 442:333-343.
- L. H. Leenheers, R. Engel, W.E.T Spruit, W.J.A. Meuling, M.J.M. Jongen (1993) Determination of methy1 5-hydroxy-2-benzimidazole carbamate in urine by high-performance liquid chromatography with electrochemical detection. J. Chromatogr. 613:89-94.
- K. Sato, J.-Y.Jin, T.Takeuchi, T.Miwa, K.Suenami, et al. (2001) Integrated pulsed amperometric detection of glufosinate, bialaphos and glyphosate at gold electrodes in anion-exchange chromatography. J. Chromatogr. 919:313-320.
- M. del Pilar da Silva, J.R. Procopio, L. Hemjndez (1993) Evaluation of the capability of different chromatographic systems for the monitoring of thimerosal and its degradation products by high-performance liquid chromatography with amperometric detection. J.Chromatogr. 653:267-273.
- S.H. Kang, H. Kim (1997) Simultaneous determination of methylparaben, propylparaben and thimerosal by high-performance liquid chromatography and electrochemical detection. J. Pharm. Biomed. Anal. 15:1359-1364.
- A. Laganil, A. Marino (1991) General and selective isolation procedure for high-performance liquid chromatographic determination of anabolic steroids in tissues. J. Chromatogr. 588:89-98.
- Th. Reuvers, E. Perogordo, R. Jim6nez (1991) Rapid screening method for the determination of diethylstilbestrol in edible animal tissue by column liquid chromatography with electrochemical detection. J. Chromatogr. 564:477-484.
- M.D. Rose, G. Shearer (1992) Determination of tranquilisers and carazolol residues in animal tissues using high-performance liquid chromatography with electrochemical detection. J. Chromatogr. 624:471-477.
- K.Lee, K. Dabrowski (2002) High-performance liquid chromatographic determination of gossypol and gossypolone enantiomers in fish tissues using simultaneous electrochemical and ultraviolet detectors. J. Chromatogr. 779:313.
- E. Kirchmann, R.L. Earley, L.E. Welch (1994) The electrochemical detection of penicillins in milk. J. Liquid Chromatogr 17:1755-1772.
- C.O.Dasenbrock, W.R.LaCourse (1998) Assay for cephapirin and ampicillin in raw milk by high performance liquid chromatography-integrated pulsed amperometric detection. Anal. Chem. 70:2415-2420.
- T. Galeano Dfaz, A.Guiberteau Cabanillas, M.I.Acedo Valenzuela, C.A.Correa, F. Salinas (1997) Determination of nitrofurantoin, furazolidone and furaltadone in milk by high-performance liquid chromatography with electrochemical detection. J. Chromatogr. 764:243-248.
- T.Galeano-Dfaz, A.Guiberteau-Cabanillas, N. Mora-Diez, P.Parrilla-Vazquwez, F.Salinas-Lopez. (2000) Rapid and sensitive determination of 4-nitrophenol, 3-methyl-4-nitrophenol, 4, 6-dinitro-o-cresol, parathion-methyl,fenitrothion, and parathion-ethyl by liquid chromatography with electrochemical detection. J. Agr. Food Chem. 48:4508-4513.





