A Description on Study of Intestinal Barrier, Drug Permeability and Permeation Enhancers
Chauhan NS1*, Alam S2, Mittal A1, Bajaj U3
1 Department of Pharmaceutics, KIET School of Pharmacy, Ghaziabad, India
2 Department of Pharma-Chemistry, KIET School of Pharmacy, Ghaziabad, India
3 Department of Pharmacology, KIET School of Pharmacy, Ghaziabad, India
*Corresponding Author
Nitesh S Chauhan,
Department of Pharmaceutics,
KIET School of Pharmacy,
Ghaziabad, India
Tel: +91-9999279008
E-mail: nschauhan84@gmail.com
Article Type: Research Article
Received: June 27, 2013; Accepted: July 25, 2013; Published: July 27, 2013
Citation: Chauhan NS, Alam S, Mittal A, Bajaj U (2013) A Description on Study of Intestinal Barrier, Drug Permeability and Permeation Enhancers.Int J Clin Pharmacol Toxicol. 2(5), 73-81. doi: dx.doi.org/10.19070/2167-910X-1300015
Copyright: Chauhan NS© 2013. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
Abstract
Penetration enhancers are present in a large number of transdermal, dermatological, and cosmetic products to aid dermal absorption of curatives and aesthetics. Delivery of hydrophilic drug by per oral route has always fascinated the researchers. Hydrophilic drugs show low bioavailability through oral administration because of their poor intestinal permeation & absorption. In the last few years there has been great interest focused on search of different intestinal permeation enhancers for the oral delivery of BCS class III drugs, small polar molecules, vaccines, hormones, peptides and proteins, which are well suitable for the delivery of the above products to give enhanced bioavailability by increasing intestinal permeability. This review targets to discuss functioning, anatomy and physiology of the intestinal barrier, drug absorption from intestinal tract, mechanism of intestinal drug permeability, detail information about intestinal permeation enhancers and its mechanism of action, in vitro methods for studying drug permeability, and importantly the advantages and applications of newer permeation enhancers.
2.Introduction
3.Diffusion Through Skin
4.Physiology of Barriers
4.1.Tight junctions and epithelial barrier function
4.2.Biochemical composition of the tight junctions
4.3.Regulation of tight junction permeability
4.4.Role of the perijunctional actinmyosin ring
4.5.Role of calcium
4.6.Role of CAMP
4.7.Role of ATP depletion
5.Drug Absorption from the Gastro-Intestinal Tract
5.1.Mechanisms of intestinal drug permeability
5.2.Passive transcellular transport
5.3.Active transport
5.4.Paracellular transport
6.Intestinal Permeation Enhancers
6.1.Mechanism of permeability enhancers
6.1.1.Surfactants
6.1.2.Fatty acids & its derivatives
6.1.3.Chelating agents
6.1.4.Chitosan’s & derivatives
6.2.Other enhancers
6.2.1.Zonula occludens toxin (Zot)
6.2.2.Polycarbophyl-cysteine conjugates (PCPCys)
6.2.3.Permeability enhancer safety
7.In-Vitro Methods For Drug Permeability [51]
7.1.Brush Border Membrane Vesicles
7.2.Isolated Intestinal Cells
7.3.Everted Intestinal Rings
7.4.Everted Intestinal Sacs
7.5.Cultured cells
7.6.Artificial membranes
8.Ideal Characteristics Of Intestinal Permeation Enhancers
9.Conclusion
10.References
Keywords
Intestinal Permeation Enhancers; Tight Junctions; Oral Bioavailability; BCS.
Introduction
To optimize bioavailability of orally administrated drug is one of the most important aims for the pharmaceutical research arena. Transport across mucosal membranes is a fundamental step for oral absorption and systemic availability. The drugs which are small and lipophilic in nature are easily permeated through the intestinal barrier whereas oral administration of macromolecules are restricted by the intestinal epithelial barrier which results in greatly reduced bioavailability .A great number of currently available drugs fall under the class III of the biopharmaceutical classification system [1], possess high therapeutic potential but cannot be delivered by oral route because of its poor permeation across the GIT epithelia. In general, these are hydrophilic compounds, of medium to high-molecular weight, and sometimes containing strongly charged functional groups implying that transport across the intestinal barrier occurs essentially via the paracellular pathway. The contribution of the latter to intestinal absorption is considered to be small, since this pathway occupies less than 0.1% of the total surface area of the intestinal epithelium, and the presence of tight junctions (TJ) between the epithelial cells limits drug absorption. These drugs have low intrinsic membrane permeability, probably because of their low lipophilicity and zwitterion character at physiological pH or act as a substrate to drug efflux pumps like p-glycoprotein, ionic charge and high molecular weight. WHO listed out the BCS class III drugs, they are shown in below Table-1.
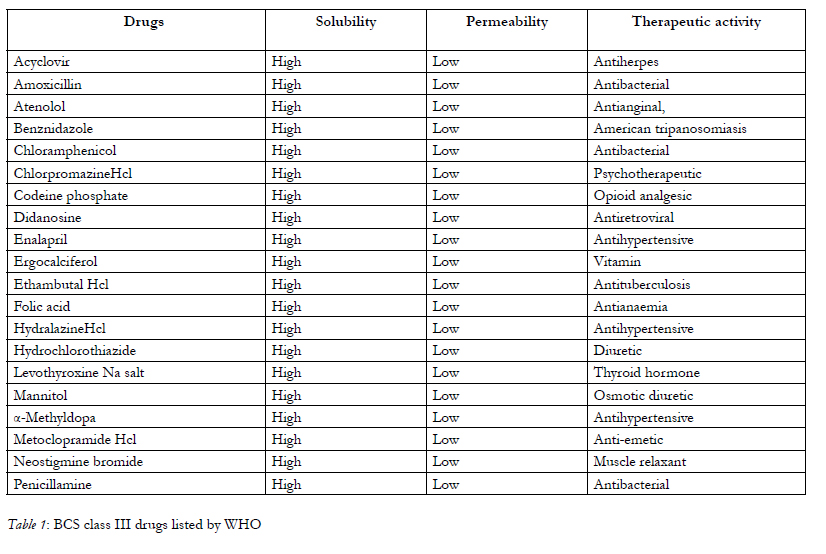

Table 1: BCS class III drugs listed by WHO.
Diffusion Through Skin
Diffusion Through Skin Fick’s 2nd Law of diffusion
dC/ dt = D d2C/dx2---------- (1)
Where,
C = Conc. Of drug
x = space co-ordinate
D = Diffusion coefficient
t = Time
Under steady state condition,
dm / dt = D C0 / h----------------(2)
Where,
m= cumulative mass of drug that passes per unit area of membrane in time t
C0 = Conc. of diffusant in the first layer of membrane at the skin surface
h = Membrane thickness
C0 = P C’0---------- (3)
Where,
P = Partition coefficient
Substituting eq. (3) in eq. (2) gives
dm / dt = D C’0 P / h----------- (4)
From equation it can be seen that :
• Diffusion coefficient of drug in stratum corneum.
• Dissolved effective conc. of drug in the vehicle.
• Partition coefficient of drug between the formulation & stratum corneum.
• Membrane thickness.
Physiology of Barriers
The barrier is composed of a single layer of columnar epithelial cells, primarily enterocytes and goblet cells, joined at their apical surfaces by tight junctions [2].
Tight junctions restrict epithelial cells immediately below the brush border forming a seal between neighbouring epithelial cells. This seal acts as a gate to restrict passage of small molecules in a charge specific manner and completely occludes diffusion of molecules with molecular radii larger than 0.1nm. In addition, the tight junction acts as a fence that separates components of the apical and basolateral domains of the epithelial plasma membrane [3].
The biochemical composition [4] of tight junctions is still being elucidated, but many of the key components have been identified. It is currently recognized that the tight junctions primarily are complex multicomponent protein structures. Identification of the principal transmembrane component has only recently come to light and it is now known that the tight junction is composed of a homotypic protein termed occludin. Other tight junctional complex proteins which have been identified are ZO-15, claudin. All of these proteins are oriented peripheral to the cytoplasmic surface of the tight junction complex and are thought to be involved in the stabilization and/or regulation of tight junction integrity.
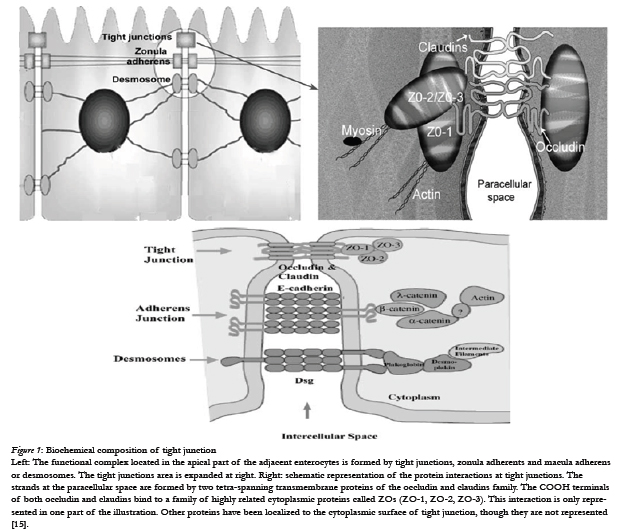
Figure 1: Biochemical composition of tight junction.
Left: The functional complex located in the apical part of the adjacent enterocytes is formed by tight junctions, zonula adherents and macula adherens or desmosomes. The tight junctions area is expanded at right. Right: schematic representation of the protein interactions at tight junctions. The strands at the paracellular space are formed by two tetra-spanning transmembrane proteins of the occludin and claudins family. The COOH terminals of both occludin and claudins bind to a family of highly related cytoplasmic proteins called ZOs (ZO-1, ZO-2, ZO-3). This interaction is only represented in one part of the illustration. Other proteins have been localized to the cytoplasmic surface of tight junction, though they are not represented [15].
The tight junction complex is not a static structural component as once was thought, but slightly resembles a dynamic and elaborate protein signalling complex. Regulation of tight junction paracellular permeability by various physiological, pathological, and experimental agents has been extensively examined in a number of in situ and in vitro culture models, particularly Caco-2 [6], brain endothelial, and MDCK cells. Peptide hormones, cytoskeleton perturbing agents, oxidants, Ca++ chelators and ionophores has all been shown to alter paracellular permeability by disrupting tight junctions. In addition, it has been determined that tight junction permeability is influenced by nearly all second messenger and signalling pathways, such as tyrosine kinases, Ca++, protein kinase C (PKC), protein kinase A (PKA), G proteins, calmodulin, CAMP, and phospholipase C.Two factors which appear to play a prominent role in the regulation of paracellular permeability by absorption enhancers are contraction of the perijunctional actinmyosin ring, and protein kinase or phosphatase mediated changes in tight junction protein phosphorylation [16].
Adjacent to the tight junction in the cytoplasm is an actinmyosin ring which restricts the cell. This ring is associated with both the tight and intermediate junctional complexes and can contract exerting an inward force on the lateral plasma membrane [17]. Such contractions are ATP-dependent and have been correlated with a loosening of the tight junctions indicating that contractions of the perijunctional ring pull on tight junction components and induce changes in paracellular permeability between neighbouring cells. Further evidence for a physical link between the perijunctional actinmyosin ring and tight junctions has been inferred by direct observation of tight junction-associated actin and by observations showing disruption in the structure and integrity of tight junctions by agents which disrupt actin filaments (e.g. cytochalasin D) possible to visualize the perijunctional actinmyosin ring by staining actin filaments with fluorescent labeled phalloidin[7]. Using this approach global changes in actin distribution have been documented with some tight junction disrupting agents including oxidants , protein kinase C activators , Ca++ depletion , and cytoskeleton disrupting agents while more subtle changes have been observed with other tight junction disrupting agents (i.e. interferon-y) [18] .
Extracellular calcium levels play a prominent role in the formation and regulation of tight junctions and paracellular permeability. Adhesion at the adherence junction is mediated by cadherin’s [7] which are Ca++ dependent, cell-cell adhesion molecules that interact homotypically. Removal of Ca++ has been known for many years to lead to an increase in tight-junction permeability and cause a redistribution of tight junction proteins. It appears that it is the disruption of cadherin adhesiveness by removal of Cat+ rather than a direct effect on the tight junction, which leads to the increase in paracellular permeability. In addition, sensitivity of cadherin adhesiveness to Ca++ can be modulated by intracellular signalling events, such as. Tyrosine phosphorylation [19].
Whereas extracellular Ca is required for formation and maintenance of tight junctions, intracellular Ca++ may be involved in regulation of tight junction permeability. In isolated hepatocyte couplets (another cell model commonly used to investigate tight junction regulation), A calcium channel blocker has been shown to increase paracellular permeability with an accompanying inhibition of intracellular calcium.
Intracellular CAMP [8] alters paracellular permeability by reducing NaCl diffusion potentials and increase passive permeability to Cl- as well as Cl: Na permeability ratios in intestinal and gall bladder epithelium. CAMP may also decrease tight junction resistance, but this effect may be masked by the increased resistance that accompanies collapse of the lateral spaces. The exact role of CAMP in regulation of tight junction is not yet clear [20].
Under normal physiological conditions, the tight junction is maintained by an energy-dependent (ATP) process involving the actin cytoskeleton and tight junctions [21]. Alteration in cellular energy status, a decrease in adenosine triphosphate (ATP) levels, has been shown to disrupt epithelial barrier function and increase permeability. Energy depletion results in net loss of phosphorylation of brush border, and possibly junctional, proteins.
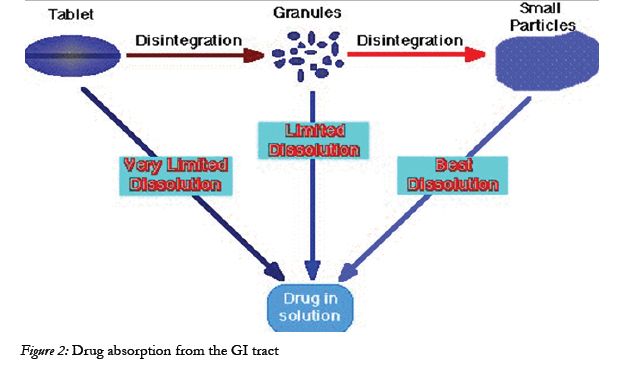
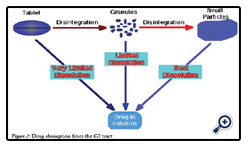
Figure 2: Drug absorption from the GI tract.
Drug Absorption from the Gastro-Intestinal Tract
Drug absorption following oral administration is a fairly complex Depending on the physicochemical properties of the drug, dissolution rate or the transport rate across the intestinal epithelium may be the rate limiting step for drugs to enter the systemic circulation [22].
The intestinal mucosa can be considered as a system of sequential barriers to drug absorption, the outermost barrier being the mucus layer and the unstirred water layer. The gel-like structure of the mucus is thought to be a sequential series of events outlined in the given figure 3 below [23].
Barriers to absorption of highly lipophilic drugs and some peptides because of the restricted diffusion in this matrix. The absorptive epithelium lining the GI tract follows the folds and villi that increase the anatomical surface area of the mucosa several-fold in the small intestine. The villi are interspaced with crypts in which the regeneration of intestinal cells occurs. In between the crypts and the tips of the villi are the basal parts of the villi. The properties which are relevant for drug absorption differ between the cells along the crypt-villus axis [9].
The main purpose of the intestinal epithelium is not only to restrict access and in this way protect the body from harmful agents, but also, to allow selective absorption of nutrients and secretion of waste products and xenobiotic [24] Schematic illustration of the different transport routes that are relevant for drug absorption [25]
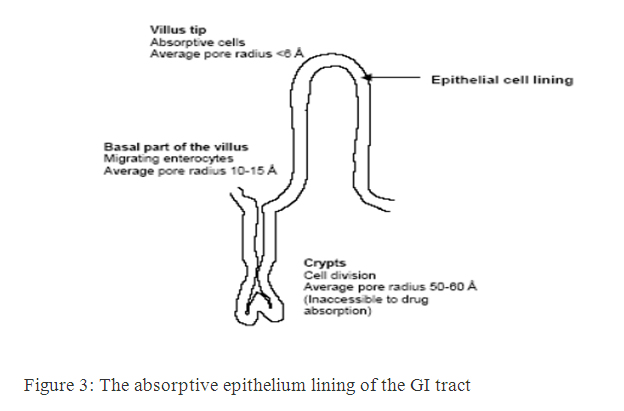
Figure 3: The absorptive epithelium lining of the GI tract.
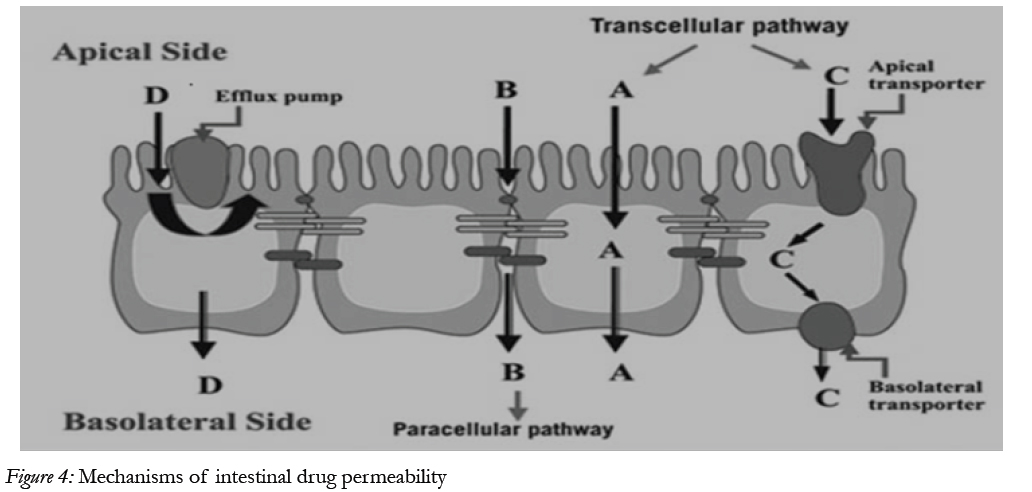
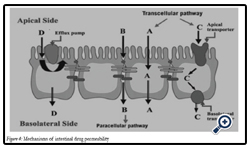
Figure 4: Mechanisms of intestinal drug permeability.
Drug transport via the passive transcellular route requires that the solute permeates the apical cell membrane. Cell membranes are made up of phospholipids arranged in bilayers that are intermingled with membrane proteins. The composition of phospholipids and proteins varies from cell type to cell type and may theoretically give rise to different permeability properties depending on the cell type. In addition, the intestinal enterocytes have a polarized cell membrane with distinct differences in membrane composition in the apical and the basolateral membrane [26]. It is generally believed that the apical membrane has a lower permeability than the basolateral membrane and the former is therefore considered to be the rate limiting barrier to passive transcellular drug transport.
Transport proteins embedded in the apical cell membrane actively shunt nutrients such as peptides, amino acids and sugars across the phospholipid bilayer. In order to restrict access of unwanted solutes via this pathway, these transporters display substrate specificity. Therefore, in order to utilize this pathway to increase absorption, the drug has to share some structure similarity with the normal substrate. A limited number of drugs are substrates for uptake carrier proteins. These include some cephalosporin antibiotics, cytostatic and angiotensin-converting enzyme (ACE) inhibitors that are substrates for oligopeptide transporters. The oligopeptide transporters have unusually broad substrate specificity, are abundantly expressed in the small intestine [38] and have therefore been the deliberate target for redesigning pharmaceuticals such as antiviral drugs to make them substrates for this transport protein [27]. Common to all absorption processes involving transport proteins is that they are saturable. Drugs that are substrates for an active transport protein can therefore display a non-linear dose-response relationship resulting in a decreasing absorbed fraction with an increasing dose. In addition, these proteins are transporters of nutrients, and therefore their capacity is likely to be influenced by food intake.
These factors may complicate the oral delivery of drugs that are absorbed by active mechanisms [28]. In contrast to transport proteins acting in the absorptive direction, the active efflux proteins secrete certain drugs that are substrates for these efflux proteins. The most well-studied efflux proteins belong to the adenosine triphosphate (ATP)-binding cassette (ABC) superfamily of membrane transporters. These include the multi-drug resistance [1] (MDR1; ABCB1) gene product P-gp and the multi-drug resistance protein family (MRP; ABCC). More recently the breast cancer related protein (BCRP, ABCG2) has been identified as potential contributor in actively limiting oral bioavailability of some drugs. The function of the efflux proteins in the intestine may be to prevent the uptake of toxic substances and also, to eliminate such substances from the blood [29].
Drugs of small to moderate molecular weights (MWs) can permeate the intestinal epithelium through the water-filled pores between the cells. This process is known as paracellular transport, and is generally considered to be a passive process, even if this pathway appears to be selective for cationic rather than anionic and neutral drugs [30]. The paracellular pathway has also been shown to be saturable, by at least two separate mechanisms, one of which involves an intracellular process. The paracellular permeability is dynamically regulated and varies both along the path of the intestine and along the crypt-villus axis. The average pore radius of the human small intestine is 8–13 Å, which will limit the paracellular permeability of drugs >4 Å and restrict those >15 Å. The colon is even more size-discriminating since the paracellular pathway restricts drugs< 3.5Å10
Intestinal Permeation Enhancers
These are the excipients which increase the intestinal permeability of poorly absorbed drugs in the small intestine and improve the oral bioavailability. These substances promote the permeability of poorly permeable drugs mainly by opening the tight junctions, leading to the increased paracellular permeability [31].
Disruption of intestinal epithelial cell membrane leads to increase in the permeability of drugs that cross the intestinal barrier through transcellular mechanism[32].
Based on the research conducted in the last decade it has become clear that several sodium salts of medium chain fatty acids are able to enhance the paracellular permeability of hydrophilic compounds Among these MCFAs, sodium caprate is the most extensively studied and the only absorption enhancing agent included in a marketed drug product. It is added in a suppository formulation intended for human use in Sweden and Japan. In Vitro and In Situ Studies of Sodium Caprate produced information regarding its mechanism which is shown below [34].
Sodium caprate is able to modulate paracellular permeability by increasing intracellular calcium levels through the activation of phospholipase C in the plasma membrane, as represented in the above Figure. The increase in calcium levels is considered to induce the contraction of calmodulin-dependent actin microfilaments,resulting in increased paracellular permeability [36].
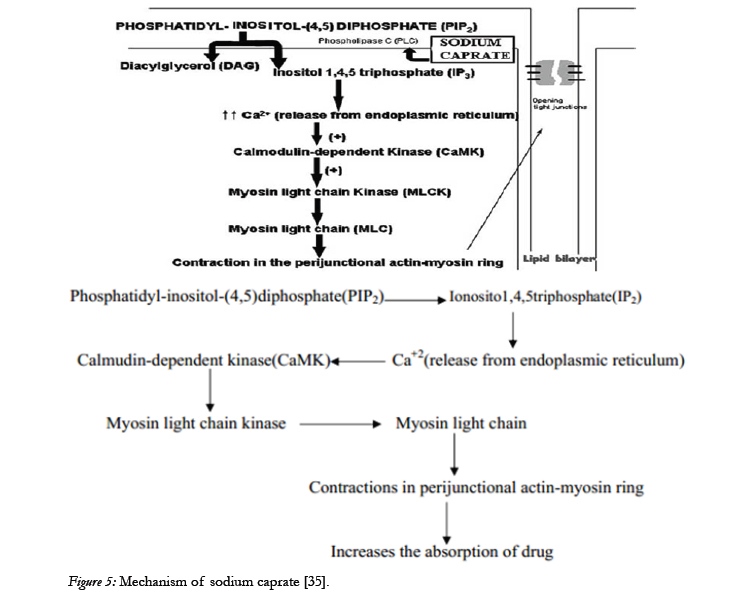
Figure 5: Mechanism of sodium caprate [35].
Chelating agent forms complexation of calcium and magnesium ions present in between intestinal epithelial cells and ultimately leads to opening of tight junctions and thereby increasing permeability for exogenous substances.
Chitosan [11] is a cationic polysaccharide obtained by partial alkaline N-deactivation of chitin. Chitin is insoluble in alkaline pH and neutral values where as its derivatives are soluble at these PH. High MW polymers such as chitosan and its derivatives have gained considerable attention as permeation enhancers. Because of their high MW, these polymers are supposedly not absorbed from the gut, and systemic side effects are thus excluded. These polymers were able to bind tightly to the epithelium and to induce redistribution of cytoskeleton F-actin and the TJ protein ZO-1, this being followed by enhanced transport via the paracellular pathway. Chitosan and its salts also act on tight junction and reduce its integrity and increases intestinal permeability. Chitosan derivatives are especially effective in enhancing the transport of small hydrophilic compounds (e.g., Mannitol) though they also improve the transport of large molecules (drugs) such as buserelin, insulin, DGAVP and octreotide acetate [37].
Zonula occludens toxin (Zot), a protein elaborated by Vibrio cholera that is able to reversibly regulate tight junction permeability. This toxin interacts with a specific intestinal epithelial surface receptor, with subsequent activation of a complex intracellular cascade of events that regulate tight junction permeability. It was also shown that the in vitro permeabilities of drugs with low oral bioavailability such as paclitaxel, acyclovir, and cyclosporine and enamione anticonvulsants were increased with Zot [38].
It is a class of permeation enhancers is represented by thiolated polymers [12] also called thiomers. These are polymers in which the thiol groups are covalently bound. It has been shown that polycarbophyl polymers (PCP) display permeation enhancing effects. This property is significantly improved as a result of the covalent attachment of cysteine (Cys) to this polymer (PCP-Cys).This thiolated polymer (PCP-Cys) is able to significantly increase the transport of marker compounds (sodium fluorescein) and peptide drugs (bacitracin-fluorescein isothiocyanate and insulin-fluorescein isothiocyanate) across the intestinal mucosa of guinea pigs. The thiol groups, covalently attached to the polymer, seem to be responsible for the improved permeation-enhancing properties of these conjugates. These compounds exert their permeation enhancing effects via glutathione [39]. It seems that PCP-Cys can transform oxidized glutathione (GSSG) to reduced glutathione 442(GSH), prolonging GSH concentration at the apical membrane. GSH is reportedly capable of inhibiting protein tyrosine phosphatase (PTP) activity by almost 100%, which leads to more phosphorylated occludin and to more open TJ as shown in the below figure [40].
The safety of absorption enhancers depends on the mechanism of action. Some enhancers may reversibly ‘loosen’ tight junctions, or transiently increase membrane permeability - without damage - under ‘very controlled’ conditions[41].
Several marketed products containing proven absorption enhancers. Yet none have reported in an increased incidence of systemic toxicity.
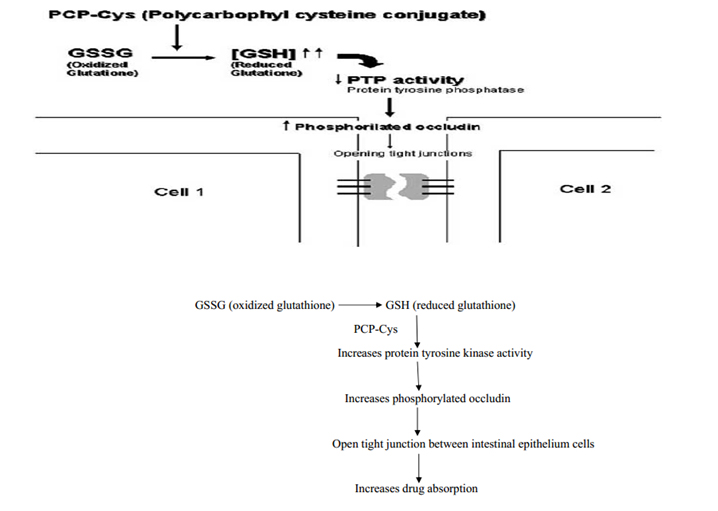
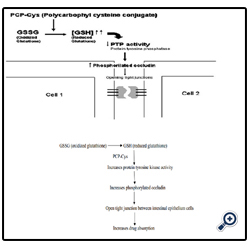
Figure 6: Mechanism action of polycarbophyl-cysteine conjugates (PCP-Cys).
In-Vitro Methods For Drug Permeability [51]
• Brush Border Membrane Vesicles
• Isolated Intestinal Cells
• Everted Intestinal Rings
• Everted Intestinal Sacs
• Cultured Cells(Caco-2 cell monolayers)
• Artificial membranes
For reasons of safety and cost, drug absorption studies [13] in humans are only carried out for a limited number of well characterized drugs. Studies of drug absorption in the intestine traditionally been carried out in experimental animals. However, the introduction of combinatorial chemistry and high throughput pharmacological screening in drug discovery has significantly increased the number of compounds entering the pre-clinical phase, and this has impossible to assess the absorption properties of all these compounds in experimental animals. This fact has spurred the development and use of vitro methods to assess drug permeability properties in most drug discovery settings. Also, the insight that drug absorption across biological barriers is a complex process involving several pathways that cannot easily be delineated in experimental animals has resulted in the large interest in academic and industrial institutions in these methods .The methods are, cultured cells and artificial membranes [43].
In this approach, cell homogenates or intestinal scrapings are treated with the CaCl2 precipitation method using centrifugation. The final pellet contains the luminal wall-bound proteins and phospholipids, which contain most of the brush border enzymatic and carrier activity. Resuspension of the pellet in buffer results in the formation of vesicles. These vesicles are mixed with the permeant in buffer and filtered after a fixed time; the amount of permeant taken up by the vesicles is then determined. Because the precipitation– centrifugation procedure results in isolation of only the brush border components, typically only the apical transcellular transport is measured by this system.
Isolated cells from the intestine of animal or human origin can be used as uptake systems in the assessment of oral bioavailability and have been described in several papers. The procedures used to isolate mucosal cells can be divided fundamentally in two categories: an in situ procedure, in which the intestine is perfused with enzyme solutions that release the cells; and an ex vivo approach, in which the cells are treated by chelating agents or by enzymatic means . The freshly isolated cells are immediately suspended in Krebs–Henseleit buffer with 10 mM glucose added and kept on ice for 15 min, during which they are bubbled with carbogen (95% O2/5% CO2). The exposure to glucose increases the viability of the cells, even after the media have been replaced by glucose-free media. In a typical experiment, the cells are separated from the primary buffer by centrifugation, resuspended in buffer under O2/CO2 in the presence of the permeant, and shaken well. After a designated time, the cells are separated by gradient-centrifugation or rapid filtration, and extracted.
The use of intestinal rings for absorption measurement is not widespread, but still has its merits in terms of simplicity. In this method, a section of the intestine is isolated immediately after euthanizing the animal, washed in icecold buffer to remove debris and digesNitesh S Chauhan, International Journal of Clinical Pharmacology & Toxicology 2013, 2:501 10 tive products, and tied at one end with a piece of suture; now, the closed end is carefully pushed through the intestine using a glass rod, resulting in eversion of the intestine, which is then cut into small rings, typically 2–4 mm wide (30–50 mg wet weight). Any segment of the intestine can be selected, from the duodenum to the rectum, typically jejunum, ileum, and colon. Next the slices are incubated in a solution containing the compound under investigation, and shaken well in a waterbath. After a designated time interval the tissue slice is taken out of solution, blotted dry, and weighed, and then dissolved or processed for assay. The uptake of the compound is measured by radiolabel counting or other means.
In the everted intestinal sac method, a 2- to 4-cm section of the intestine is tied off at one end and everted using a glass rod or a thread, similar to the procedure described for the intestinal rings . As with the everted slices and rings, the mucosa becomes the outer side of the sac and is in contact with the incubation medium, but in contrast with the rings, only the mucosa is in contact with permeant. The sac is filled with buffer and put in a flask with oxygenated (95% O2/5% CO2) buffer containing the compound under investigation. At the end of the experiment the sac is cut and opened at one end, and the serosal fluid is collected . Viability of the sac can be monitored during the experiment by measuring the transport of a marker (e.g., trypan blue dye).
The human adenocarcinoma Caco-2 [14] model suitable for screening intestinal drug permeability and predicting the oral absorption potential of new drug substances. The Caco-2 cells were grown on permeable supports and spontaneously formed polarized monolayers that resembled that of the intestinal epitheliums shown in below figure.
In many respects the Caco-2 cells are therefore functionally similar to the human small intestinal enterocyte, despite the fact that they originate from a human colorectal carcinoma. The methods that are based on Caco-2 are, however, not only useful for drug absorption screening. It is also extract information about specific transport processes that would be difficult to obtain in more complex models such as those based on whole tissues from experimental animals. For methods enable us to investigate the relative contribution of passive transcellular and paracellular transport, the effect of charge on paracellular transport and the effect of solvent drug. Caco-2 is the most widely used cell line for drug permeability studies [44].
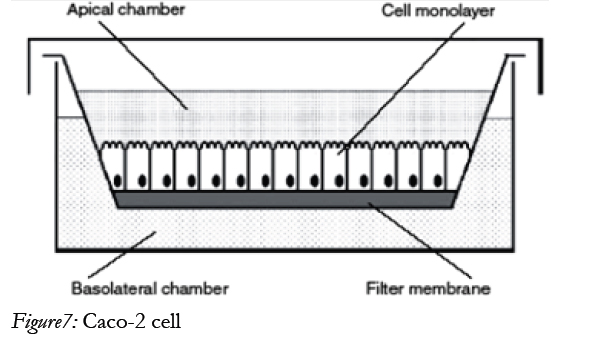
Figure7: Caco-2 cell.
• Epithelial cell cultures.
• Usage of immobilized phospholipids or liposome in organic solvent.
• Chromatographic methods where the stationary phase consists of immobilized phospholipids or liposome.
These methods are attractive for screening purposes since they require very little compound, are easily automated and are adaptable to diverse sets of drugs [45].
Ideal Characteristics Of Intestinal Permeation Enhancers
• Non toxic, non irritating, non allergic.
• Ideally work rapidly.
• Pharmacologically inert.
• Its duration of action should be predictable & reproducible.
• Should work unidirectionally.
• When removed from skin barrier properties should return both rapidly & fully.
• Cosmetically acceptable.
• Compatible with both excipients & drug.
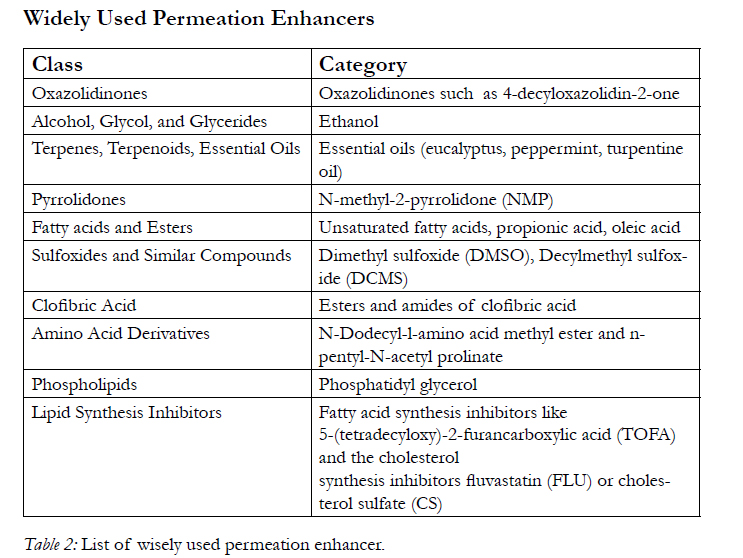
Table 2: List of wisely used permeation enhancer.
Conclusion
It is most desirous that an absorption enhancer assist the transport of drug via intestinal barrier to increase the bioavailability of administered drug. By using of different intestinal permeation enhancers, drugs having poor bioavailability could be fabricated in such a novel way. Fortunately apparently safe and effective intestinal permeability enhancers are currently available, so formulation expertise can make best use of enhancer while formulating novel drug delivery system. This extensive review provides comprehensive details on various permeability enhancers used in delivering drug content through membrane barrier.
References
- H. Hidaka, M.inagaki, S. Kawamoto and Y. Sasaki, Biochemistry.23,5036 (1984)
- H. Itoh and H. Hidaka, J Bioche. 96, 1721 (1984)
- S. Soboll, S. Gru¨ ndel , U. Schwabe and R.Scholz, Eur J Biochem.141, 231(1984).
- JL. Madara, D. Barenberg and S. Carlson, J Cell Biol. 102, 2125,(1986).
- M. Saitoh, T. Ishikawa, S. Matsushima, M. Naka and H. Hidaka, J Biol Chem. 262, 7796, (1987).
- JL. Madara, J. Stafford, D. Barenberg and S. Carlson, Am J Physiol.254, G416, (1988).
- D. Hollander, D. Ricketts, CAR. Boyd and D. Phil, Can J Gastroenterol.2, 35A, (1988).
- P. Artursson, J Pharm Sci. 79, 476, (1990).
- MS. Balda, J Cell Biol. 122:193, (1991).
- PE. Canfield, AM. Geerdes and BA. Molitoris, Am J Physiol. 261,F1038, (1991).
- P.Artursson and J. Karlsson, Biochem Biophys Res Commun. 175, 880,(1991).
- T. Sawada, T.Ogawa, M. Tomita, M. Hayashi and S. Awazu, Pharm Res. 8, 1365, (1991).
- EK. Anderberg, C. Nystro¨m and P. Artursson, J Pharm Sci, 81, 879,(1992).
- B. Ellis, EE. Schneeberger and CA. Rabito, Am J Physiol, 263, F293, (1992).
- MH. Nathanson, A. Gautam, OC. Ng, R. Bruck and JL.Boyer, Am J Physiol, 262, G1079, (1992).
- DI. Yule and JL. Williams, J. Biol. Chem. 267, 13830, (1992).
- MG. Tansey, RA. Word, H. Hidaka, HA. Singer, CM. Schworer, KE.Kamm and JT. Stull, J Biol Chem. 267, 12511, (1992).
- MS. Balda, L. Gonza´lez-Mariscal, K. Matter, M. Ceredijo and JM.Anderson, J Cell Biol. 123, 293, (1993)
- EK, T. Lindmark and P. Artursson, Pharm Res. 10, 857, (1993).
- JM. Mandel, R. Bacallo and G. Zampighi, Nature. 361, 552, (1993).
- H. Schulman, Curr Opin Cell Biol. 5, 247, (1993).
- WF. Stenson , RA. Easom, TE. Riehl and J. Turk, Am J Physiol. 265,G955, (1993)
- P. Artursson , T. Lindmark, SS. Davis and L. Illum, Pharm Res. 11,1358 (1994).
- R. Bacallo, A. Garfinkel, S. Monke, G. Zampighi and LJ. Mandel. J Cell Sci. 107, 3301 (1994).
- JH. Hochman, JA. Fix and EL. LeCluyse, J Pharmacol Exp Ther.269, 813 (1994).
- MP. Walsh, Mol Cell Biochem. 135, 21, (1994).
- T. Lindmark, T. Nikkila and P. Artursson, J Pharmacol Exp Ther.275, 958 (1995).
- KL. Madsen, NL. Yanchar, DL. Sigalet, T. Reigel and RN. Fedorak,Gastroenterology. 109, 107 (1995).
- RO. Stuart and SK. Nigam, Proc Natl Acad Sci, USA. 92, 6072 (1995).
- M. Tomita, M. Hayashi and S. Awazu, J Pharmacol Exp Ther. 272,739 (1995)
- AL. Salzman, JM. Menconi, N. Unno, RM. Ezzell, DM. Casey, PK.Gonzalez and MP. Fink, Am J Physiol, 268, G361 (1995).
- W-C Yen and VHL Lee, J Pharmacol Exp Ther, 275, 114 (1995).
- YH. Tai, J. Flick, SA. Levine, JL. Madara, GWG. Sharp and M.Donowitz, J Membr Biol, 149, 71 (1996).
- NGM. Schippe, KM. Vårum and P. Artursson, Pharm Res, 13, 1686(1996).
- H. Ghandehari, PL. Smith, H. Ellens, P-Y. Yeh and J. Kopecek, JPharmacol Exp Ther 280, 747 (1997).
- T. Lindmark, JD. So¨derholm, G. Olaison, G. Alva´n, G. Ocklindand P. Artursson, Pharm Res, 14:930 (1997)
- T. Lindmark, N. Schipper, L. Lazorova´, AG. de Boer and P. Artursson, J Drug Target, in press. (1997).
- Y. Murata, N. Sasaki, E. Miyamoto, S. Kawashima, Eur.J.Pharm Biopharm 50, 221 (2001).
- R. Neubert, Y. Mrestani, C. Mrestani-Klaus, European Journal of Pharmaceutics and Bio pharmaceutics. 58, 653 (2004)
- Grassi Mario, Grassi Gabriele, Current drug delivery 2, 97-116 (2005).
- Keller TCS and Mooseker MS.; 1982 Ca11-calmodulin-dependent phosphorylation of myosin, and its role in brush border contraction in vitro. J Cell Biol, 95:943–959.
- Lee Seulki, Kim Sang Kyoon, Journal of pharmaceutical sciences,94,2541 (2005).
- P. Smrdel, Drug Development and Industrial Pharmacy. 32, 623(2006).
- L.Patel Yagnesh, AAPS. 7, E1 (2006).
- M. Harrish Shoaib, Jaweria Tazeen, Hamid A.Merchant, Pakistan Journal of Pharmaceutical Sciences. 19, 119 (2006).
- KC. Ofokansi, Adikwu and Okore, Drug Development and Industrial Pharmacy.33, 691 (2007).
- M. Simonoska, Goracinova, European Journal of Pharmaceutics and Biopharmaceutics. 68, 565 (2008).
- Bock Udo, Hiller Christian and Balser Sigrid, European journal of pharmaceutics and Biopharmaceutics. 68, 390 (2008)
- P. Anilkumar, A.V. Badarinath, N. Naveen, K. Prasad, B. Ravi Shankar Reddy, M. Hyndhavi, and M. Nirosha, Journal of Global Trends in Pharmaceutical Sciences. 2, 4, 431 (2011).





