Pretreatment, Analysis, Identification and Preparation of Toxic Compounds Eluted into Body Fluids from Medical and Dental Devices
Hideharu Shintani*
Faculty of Science and Engineering, Chuo University, 1-13-27, Kasuga, Bunkyo, 112-8551, Tokyo, Japan.
*Corresponding Author
Hideharu Shintani,
Faculty of Science and Engineering,
Chuo University, 1-13-27, Kasuga,
Bunkyo, 112-8551, Tokyo, Japan.
Tel: +81425922336, FAX: +81425922336.
E-mail: shintani@mail.hinocatv.ne.jp.
Article Type: Review Article
Received: August 23, 2014; Accepted: September 30, 2014; Published: October 01, 2014
Citation: Hideharu Shintani (2014) Pretreatment, Analysis, Identification and Preparation of Toxic Compounds Eluted into Body Fluids from Medical and Dental Devices. Int J Bioanal Methods Bioequival Stud. 1(1), 1-15. doi: dx.doi.org/10.19070/2470-4490-140001
Copyright: Hideharu Shintani © 2014. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
Abstract
Selective analysis of toxic compounds in complicated matrix such as body fluids is extremely difficult. This is because unexpected interference admixtures with almost identical retention time in chromatography may co-elute with overlapping. 1t is essential to remove admixtures byappropriate pretreatment procedures. For that purpose there may be several sorts of pretreatment methods such as liquid-liquid extraction or solid phase extraction (SPE) using column, membrane dialysis filtration ultra-filtration super fluid critical extraction SFE or adsorption using charcoal or other appropriate adsorbents. It has been reported several pretreatment methods for acidic, basic and neutral compounds in biological fluids. Body fluids direct injection column is also available in the market, however this column has restriction of concentration of organic solvent in the eluent up to 20% of methanol (18% of acetonitrile), thus resulting in poor or no elution of strongly hydrophobic compounds on reverse phase. The reason of restriction is prevention of coagulation of denaturated protein in the reversed phase column. Artifact formation must also be considered during pretreatment methods. Artifact formation may cause lower recovery of the compound of interest and may also cause misunderstanding to the researcher, which they may extract unexpected highly strong toxic compound. According to the recent advancement of analytical column fabrication technology, several new columns based on innovated technology are now commercially available. Some of them can be resulted in incompletely residual silanol column, so common ion addition is required for strongly basic or acidic compounds analysis or ion-pair method is required. Ion-pair method is so popular but it often causes to shorten column life by retaining and spoiling the column with ion paring reagent. These columns are also described by comparing conventional columns in this paper.
2.Introduction
2.1.MDA
2.2.Toxic compounds from dental material
2.3.Urea and Uric acid
2.4.DEHP
2.5.bisphenoI A
2.6.Bisphenol S and 4-chloro-4’-hydroxydiphenyl sulfone
3.Selective retention, removal and elution for analysis of toxic compounds to human health
3.1.Blood MDA
3.2.Residual toxic compounds in blood from PMMA dental materials
3.3.Blood urea-pretreatment and analytical conditions
3.4.Pretreatment of PA, MEHP and DEHP in human serum and plasma
3.5.Pretreatment of bisphenol A in blood
4.SPE vs liquid-liquid extraction and the formation of artifact with solvent extraction
5.Conclusions
6.References
Keywords
Solid phase extraction Liquid-liquid extraction 4; 4'-methylenedianiline Dental material Urea; 2-diethylhexyl phthalate; Bisphenol A; HPLC analysis; ECD detection; UV detection; Body fluids; Cytotoxicity; Artifact
Introduction
Solid phase extraction SPE using packed columns combined with high performance liquid chromatography HPLC for analysis of several toxic compounds in body fluids such as blood, urine or saliva was described. SPE and liquid-liquid extraction were also compared in terms of recovery efficiency, solvent consumption handling time artifact formation and so on. Membrane type SPE for large scale extraction especially applicable for environmental analysis will also be described. Membrane type SPE was not successfully applicable to biological fluids due to mostly stacking of membrane pore, therefore so often restricted to environmental analysis treatment in such a case of analyzing trace residue of pesticide in water.
In this paper the author will describe major six substances from his published papers in past. They are 4,4’-methylenedianiline MDA from γ-ray irradiated polyurethane PU for sterilization purpose residue of toxic compounds from dental materials blood urea as uremic toxins endocrine disrupters of DEHP (2-diethylhexyl phthalate and bisphenol A and bisphenol S and 4-chloro4’-hydroxydiphenyl sulfone.
The first compound is MDA [1-7]. As a medical use thermosetting PU was mainly used. For example, potting material for connecting dialysis fibers with housing of an artificial dialyzer was also made from thermosetting PU. In fabrication of thermosetting PU, polyol, methylenediisocyanate (MDI) and butanediol were mixed and terminated polymerization with addition of butanol to obtain an appropriate molecular weight of PU. Residual MDI changed to MDA by hydrolysis and also MDA produced by cleavage at urethane linkage by irradiation for sterilization [1-7]. MDI is more toxic and reactive than MDA. MDA in blood was analyzed to assess human risk contacted with PU medical devices during artificial dialysis to patients [1-7]. MDA production from γ-ray irradiation of PU is the first finding by the author. MDA was determined by reversed-phase HPLC using an eluent of a mixed solution of ammonium acetate and acetonitrile at a ratio of 7/3 (v/v) [8-13]. MDA was reported to be unstable upon heating,therefore HPLC determination is superior to gas-liquid chromatography (GLC) due to free from high temperature heating. At that time when the author reported MDA determination [8-11], the conventional endcapped C-18 column only was available in the market. Following the conventional endcapping procedure it remained many untreated residual silanol (around 80%) which indicates an incomplete endcapping. So, complete endocapped column is desired. This will be written later. Detection was by an electrochemical detector (ECD amperometry ICA-3000R from Towadenpa Co., Tokyo) as well as ultraviolet (UV) detection at 235 nm. ECD was more sensitive and selective than UV detection however ECD detection was restricted to the compounds with relatively lower oxidation-reduction potential (less than 1000 mV) such as aromatic amine phenolic OH phenolic SH or aliphatic SH or OH such as cystine or sugar respectively. Baseline of UV detection was fluctuated due to impurities in the eluent however ECD detection indicated flat baseline [11]. Detection limit of MDA by UV at 235 nm and that by ECD at 900 mV was 150 and 3 ng/mL, respectively [11].
Concerning pretreatment of compounds in body fluid several methods were compared in terms of their efficiency recovery and other factors. Liquid-liquid extraction was troublesome due to necessity of deproteinization followed by centrifugation and condensation. During condensation there may be a possibility of recovery loss due to evaporation. Manual type SPE and an automated SPE were also compared in terms of reproducibility of recovery rate. Furthermore, artifact formation described later must be considered when using liquid-liquid extraction. Blood MDA was satisfactory recovered using C-18 Phenyl and Cyclohexyl SPE columns (Table l) indicating major retention mechanism may be van der Waals binding as well as Π-Π binding.
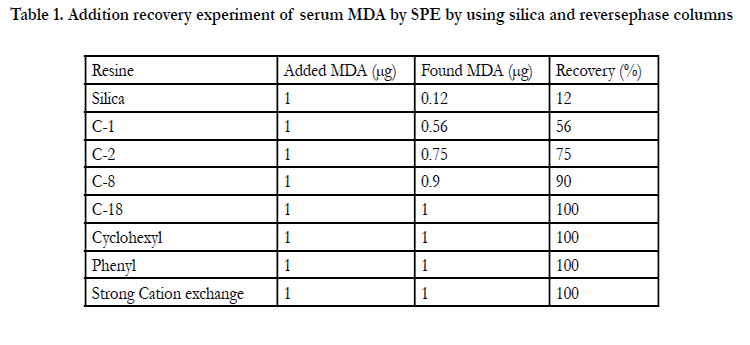

Table 1. Addition recovery experiment of serum MDA by SPE by using silica and reversephase columns
The recovery of serum MDA from C-1 C-2 C-8 and Silica was unsatisfactory (Table l).Elution was carried out using methanol containing 1M NH4OH. According to the conventional technique of SPE and reported procedure for basic compounds, it was speculated that acidified methanol (methanol containing 1M HCI) as the eluent was thought to be superior, however the experimental result showed the opposite result contrary to our speculation (Figure l). The speculated explanation for the mechanism will be described in the text. The result suggested the importance to consider simultaneously the residual silanol effect and chemical status of compound of interest. SPE was superior to a liquid-liquid extraction due to an unnecessary of deproteinization centrifugation condensation greater recovery, less consumption of organic solvent, less experimental time and less possibility of artifact formation. SPE procedure of blood MDA was described in Figure 2. The procedure in Figure 2 was carried out by using automated SPE of BenchmateR.

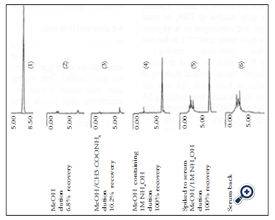
Figure 1. HPLC chromatograms of MDA treated with different eluents in SPE using C-18 column HPLC conditions: C-18 column, Eluent: methanol and an aqueous solution of 10mM ammonium acetate at a ratio of 1/1, Flow rate: 1.2ml/min, Detection: 254 nm. MDA peak has a retention time of 6.95 min.
(1) 105 ng/10 μl MDA standard solution, 10 μl applied to HPLC.
(2) 100 μl of 105 ng/10 μl MDA solution applied to C-18 resin with 100 mg and 120 μl, respectively, resin weight and void volume,and eluted with 250 μl of methanol. 200 μl collected and 10 μl applied to HPLC. When 100% is recovered, the theoretical
concentration of MDA would be 52.5 ng/10 μl.
(3) The same procedure as above expect for the elution, 250 μl of a mixed aqueous solution of 10mM ammonium acetate and
methanol at a ratio of 1/1.
(4) The same procedure as above except for the elution, 250 μl of methanol containing 1M NH4OH.
(5) 50 μl of 21 μg/ml MDA was added to 1 ml serum to prepare one μg/ml serum. This was applied to C-18 resin, eluted with
250 μl of methanol containing 1M NH4OH. 200 μl collected. Ten μl applied to HPLC. If 100% is recovered, the theoretical
concentration would be 50.0 ng/10 μl.
(6) The same procedure as above but omitting MDA, the serum blank.

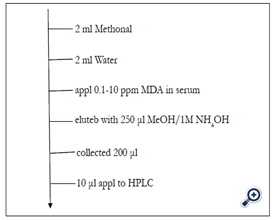
Figure 2. SPE procedure for blood MDA
C1,2, 8,18, Phenyl, Cychlohexyl and Silica (100 mg resin weight, 120 μL void volume)
Satisifactory recovery rate otained by the use C18, phenyl and cyclohexyl column
The second compound were those from polymethyl methacylate (PMMA) dental material [14-18]. PMMA is widely used as the composite resin for the dental plate. In accordance with the current PMMA fabrication benzoylperoxide (BPO) and N,Ndimethy- p-toluidine (DMPT) were added as the initiator and the stimulator, respectively, for methyl methacrylate (MMA) polymerization. If insufficiently polymerized, MMA monomer and starting compounds of DMPT and BPO exhibit a residue potential. Especially residue of MMA was significant at around l -2%.When considering 1-2%of toxic compound of MMA may be eluted into patient’s body fluids during dental treatment this amount of residue must be diminished for patient's sake. As one proposed method for diminishment was rinsed with hot water as MMA is hydrophilic. Rinsing with organic solvent such as with methanol must be avoided as MMA dental plate was discolored and deformed.
Newly found toxic compounds have been identified using HPLCmass spectrometry (MS) [18]. One of them was epoxide compound of DMPT. Epoxide compound was quite reactive to DNA so most probably mutagen and carcinogen as is ethylene oxide. BPO was quite easily converted to benzoic acid (BA) in a few seconds when contacting with DMPT or body fluids such as blood or saliva, therefore BA analysis has an identical meaning of BPO determination in body fluids. BA was not originally used for PMMA fabrication, indicating BA is a sort of artifact from BPO when contacting with body fluids. BPO is quite reactive compound and both BPO and BA are cytotoxic [17]. For determining cytotoxicity blood serum was added in cell culture medium, therefore BPO immediately transformed to BA, which was at first confirmed by the author. From the result cytotoxicity data reported as BPO is confirmed not the exact cytotoxicity data of BPO, but the data of BA [17]. The cytotoxicity data of IC50 (μg/mL) of BA and BPO using Balb 3T3 cell was 28.7 and 22 respectively indicating both are almost identical because BPO data was originally from BA.
In order to evaluate the risk factor to the recipient exposed to these compounds from dental material the authors quantitatively analyzed residual amount in composite resin using blood serum extraction [16-17]. MMA and BPO are unstable upon heating therefore HPLC is considered to be superior to GLC. Determination was carried out by HPLC combined with SPE using C-18 columns in analytical and SPE columns. The comparison of eluents of these compounds from C-18 SPE column was discussed. The comparison of SPE and liquid-liquid extraction in terms of recovery efficiency was also discussed later.
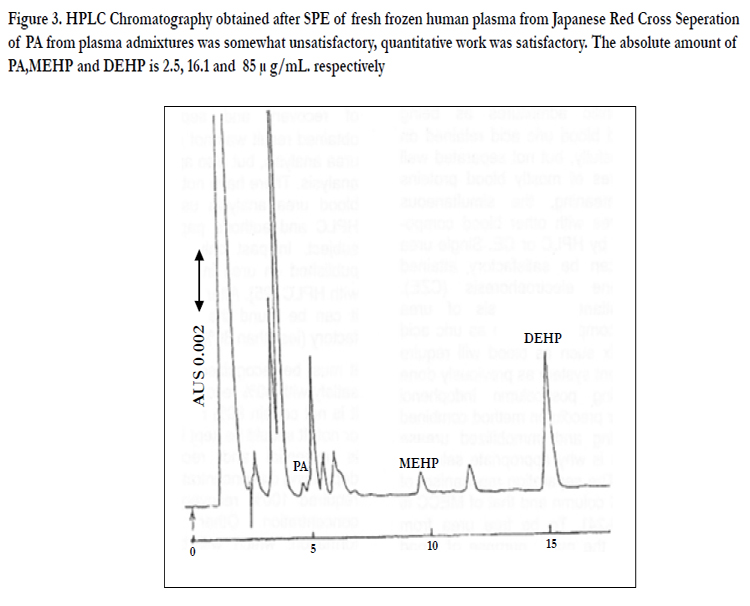
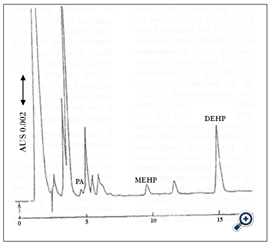
Figure 3. HPLC Chromatography obtained after SPE of fresh frozen human plasma from Japanese Red Cross Seperation of PA from plasma admixtures was somewhat unsatisfactory, quantitative work was satisfactory. The absolute amount of PA,MEHP and DEHP is 2.5, 16.1 and 85 μ g/mL. respectively
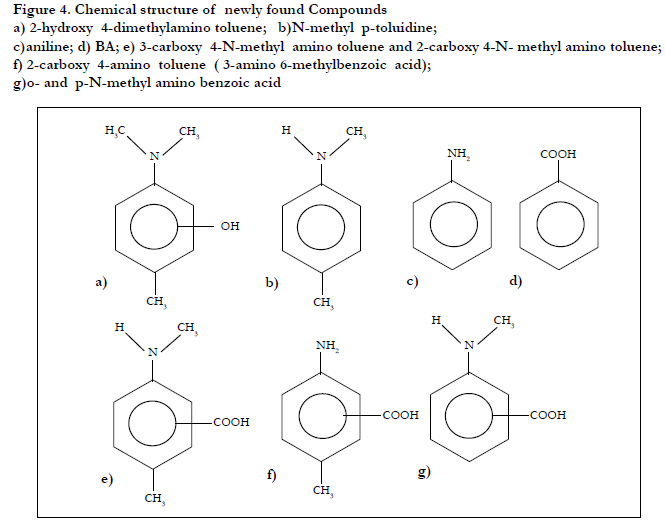
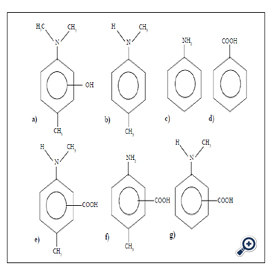
Figure 4. Chemical structure of newly found Compounds
a) 2-hydroxy 4-dimethylamino toluene; b)N-methyl p-toluidine;
c)aniline; d) BA; e) 3-carboxy 4-N-methyl amino toluene and 2-carboxy 4-N- methyl amino toluene;
f) 2-carboxy 4-amino toluene ( 3-amino 6-methylbenzoic acid);
g)o- and p-N-methyl amino benzoic acid
The third compound is blood urea and uric acid. Urea is a major uremic toxin however urea accumulation is a trigger or the result of uremia is not well understood. This means whichever blood urea may cause uremia or blood urea may accumulate after uremia promotes is currently uncertain. In healthy person protein terminally metabolites to urea as a final product through urea cycle and exclude urea to urine through kidney exclude function. Uremia patients cannot successfully exclude urea to urine as kidney function of the patient damages therefore urea accumulates back to blood. Urea is a final compound of protein in urea cycle so accumulation of final product will cause feedback inhibition, thus leading to irregular protein metabolism.
There are two types of urea. One is free from protein (mostly albumin) and the other is bound to protein type. It was reported around 3% of total urea will bind to albumin. Free urea plays an important role for promoting uremia syndrome, therefore accurate as well as differential analyses of bound from free urea in blood are important to attain precious diagnosis. Current clinical urea analysis is carried out using an ammonium selective electrode attached immobilized urease onto the electrode to determine ammonium as urea which is so-called blood urea nitrogen BUN. The alternative method for clinical test carried out at the hospital is autoanalyzer to use Indophenol colorimetry. When using these methods differential analysis of endogenous ammonium and urea was not attained. In addition differential analysis of free from bound urea cannot be attained. Autoanalyzer used in the hospital routinely determines total urea as BUN by converting urea to ammonium with urease and react with indophenol reagent to detect colorimetry with visible detection. Amount of endogenous blood ammonium can be neglected due to around l% of total blood urea, which will be within analytical error however that of urine ammonium cannot be neglected due to more than 10% of urine urea. There have been reported so far several methods for blood urea analysis [19-22].Urea analysis using capillary electrophoresis (CE) (mostly using micellar electrokinetic chromatography MECC.or MEKC) was also reported [23-24]. However, separation of urea from blood admixtures was not satisfactory as urea peak was eluted at around void volume overlapped with hydrophilic blood admixtures which was identical to urea analysis using C-18 HPLC column [19-22]. Blood urea was poorer retained on C-18 column and also eluted at around void volume overlapped with blood hydrophilic admixtures as being reported already and blood uric acid retained on C-18 column successfully. but not separated well from blood admixtures of mostly blood proteins [19-22]. In this meaning the simultaneous analysis of blood urea with other blood compounds was so difficult by HPLC or CE. Single urea analysis in blood can be satisfactory attained using capillary zone electrophoresis (CZE).However for simultaneous analysis of urea together with other compounds such as uric acid in complicated matrix such as blood will require complicated equipment system as previously conducted by the author using post-column Indophenol colorimetry method or precolumn method combined with column switching and immobilized urease column method [19-22]. This is why appropriate selection column is required. The separation mechanism of reverse phase HPLC column and that of MECC is almost identical [19-24]. To be free urea from blood admixtures is the major purpose of blood pretreatment. For selective analysis of blood urea free from blood admixtures the author discusses as follows: selection and validation of analytical condition and validation of pretreatment methods to attain selective analysis of blood urea as well as differential analysis of endogenous blood urea from endogenous ammonium. Direct blood injection is applicable to CE and HPLC however there always associates a possibility of deterioration of column and detector cell with blood protein or hydrophobic lipid as well as overlapping of compound of interest with blood admixtures [23] therefore it is more appropriate to avoid direct blood injection. Direct blood injection HPLC column has a restriction of less than 18% of acetonitrile or 20% of methanol as an eluent otherwise deproteinization will occur in the column and this may deteriorate column and detector cell, cause clogging the column and make shorten column life. The restriction of organic solvent concentration in the eluent indicates strong hydrophobic compounds cannot be appropriately eluted. When handling body fluids (blood or saliva), it is recommended to remove and isolate admixtures by pretreatment to attain significant base-line separation free from blood admixtures reproducible chromatograms and prolonged life of analytical column and detection cell.
As pretreatment methods traditional liquid-liquid extraction SPE dialysis, ultrafiltration or supercritical fluid extraction (SFE)was compared in terms of recovery and separation efficiency. The obtained result was not always restricted to blood urea analysis but also applied to blood compound analysis. There have not been published so far on blood urea analysis using SPE combined with HPLC and author's paper was the first on this subject. In past only single paper have been published on urea in food using SPE combined with HPLC [25]. After tracing the reported method, it can be found the recovery rate was unsatisfactory (less than 80%) [25].
It must be recognized that some researcher may satisfy with 80% recovery rate but problem is that it is not certain 80% recovery will be reproducible or not. It should be kept in mind that reproducibility is essential and recovery rate may differ depending on concentration. That's why the author required 100% recovery using different sample concentration. Other reason is an artifact formation which will be discussed later. The reason to be considered for the major difference of recovery rate between the author's experiment and that of Fujiwara's experiment [25] might result in different matrix of blood or food respectively. Anyhow the author studied for pretreatment method to attain 100%recovery for blood urea analysis. Differential analysis of free from bound blood urea can also be attained using ultrafiltration or dialysis for native blood. The amount of bound urea was not significant at around 3% that of total blood urea. When using denatured blood total urea amount can be determined in ultrafiltrate after ultrafiltration at around 13,000 g for 40-60 minutes. Denaturation was attained by acidification of blood to make bound urea free from protein. Endogenous ammonium in blood and urine was around l% and 10% of total urea, respectively, therefore the former was within an analytical error. As the latter is beyond the analytical error determination of endogenous urine ammonium will be required for differential analysis of endogenous urine urea.
The fourth compound is DEHP (2-diethylhexyl phthalate)in blood [26,27]. This compound is known as a carcinogen and endocrine disrupter [28]. DEHP in health care products is mainly used for the fabrication of PVC (polyvinyl chloride). Around 50% of PVC is DEHP [27], so migration of DEHP into human body must be prevented. For that purpose we need to accurately confirm how much amount of DEHP in PVC will elute into body fluids. We determine blood DEHP as well as blood MEHP (2-monoethylhexyl phthalate) and PA (phthalic acid). A pre-treatment method by automated SPE and HPLC has been developed for analysis of the endocrine disrupters of MEHP and DEHP in blood products.
For successful analysis it was necessary to use acidified blood and acidified SPE eluent to suppress ionization of MEHP and PA (ion suppression method).The amount of PA MEHP and DEHP migrated into blood products from PVC blood bags was determined [26]. It was shown that most of the MEHP detected in human plasma is not derived directly from flexible PVC bags but is produced by the action of endogenous blood lipase and esterase on the eluted DEHP [27]. The safety of PA MEHP and DEHP must be considered because it has been reported that MEHP is more toxic than DEHP [28].The method is described for simultaneous analysis of PA, MEHP and DEHP in blood with linear gradient elution after automated SPE (Figure 3)[27].
Eluent of HPLC was a mixture of acetate buffer (pH3 10 mM) and acetonitrile. The ratio of acetate buffer to acetonitrile was changed linearly from 9:l to l:9 in 17 min then changed to 100% acetonitrile in another 6 min to clean the column after which the initial eluent constituent was delivered for 5 min before start the next analysis. The total analysis time was 28 min The flow rate was l mL/min detection was at 235 nm and injection volume was 20 μL.
Suppression of ionization of carboxylate group in MEHP was essential if the compound was to be retained on the C-18 column. Thus 1:l blood and 10 mM acetate buffer pH 3 were well mixed before injection (l mL) to SPE column previously conditioned with acetonitrile (2 mL) then acetate buffer (10 mM pH3; 2 mL). The sample was then passed through the column by application of vacuum, the column was rinsed with acetate buffer (10 mM pH3; 0.5 mL) and the compounds of interest were eluted with l mL acetonitrile acidified with acetic acid at pH 2. The eluate was collected and 20 μL were injected to HPLC. Conditioning, rinsing and elution of the column were performed by using a computercontrolled SPE system of BenchMateR.
As it has been reported that conventional liquid-liquid extraction of DEHP MEHP and PA results in poor recovery [29,32] an attempt was made to develop an alternative pre-treatment method for satisfactory and reproducible recovery of these compounds in blood. Initially the author studied for the deproteinization with an acetonitrile and sodium hydroxide and then ultrafiltration, for pretreatment of serum [26,32]. The upper layer was concentrated by evaporation. Because this conventional liquid-liquid extraction procedure was complicated and tedious and the rate of recovery was varied [26,32] therefore automated SPE was studied. The automated SPE procedure was found to be simpler less time consumption and consumed less organic solvent, which is of advantage both to the analyst and the environment. The greatest benefit of using automated SPE was satisfactory recovery with sufficient reproducibility because of the availability of constant pressure control. The recovery by automated SPE was 98-102%(average 99% n=5) [27]. Manual SPE was however, not recommended because the reproducibility of recovery was in the author's experience poor between 40-105%;this indicates that accurate pressure control is essential when performing SPE.
Linear gradient elution was used for HPLC analysis after SPE (Figure 3). The retention times of PA MEHP and DEHP was 4.8 9.6 and 15.7 min respectively. There was no interference with these peaks from other compounds present in the blood indicating SPE pre-treatment was satisfactory. The recovery of PA MEHP and DEHP in blood was satisfactory. If the blood samples and the solution used to condition the SPE column were not acidified recovery of PA and MEHP from the column was poor as they were not successfully retained on the C-18 column when ionization of the carboxylate group was not suppressed. A procedure which did not use ion suppression method has been reported but the recovery of the carboxylate compounds was not satisfactory [33]. The success of the experiment was a direct result of acidification and the consequent ion suppression effect and common ion effect for acidic compounds of MEHP and PA [3,16,17,34-36]. The common ion effect must also be significant. When the SPE procedure was compared with conventional liquid-liquid extraction method SPE afforded superior recovery because the lack of need to concentrate eliminated evaporation loss [17,32,35,36]. Liquid-liquid extraction procedure requires troublesome evaporation for condensation consumption of large volumes of organic solvent and complicated handling and often results in lower recovery because of loss during evaporation [32], especially of lower boiling-point compounds. It has been reported that concentration of dioxin extracts by use of a jet of nitrogen gas resulted in lower recovery because of blowing out during procedure [32].
SEP vs liquid-liquid extraction of blood DEHP and so on will be discussed later in detail.
The fifth compound is also the endocrine disrupter of bisphenol A (BPA) [37,38]. Automated SPE then HPLC have been studied for determination of BPA in blood. Electrochemical detection was used for selective and sensitive detection of BPA. Determination of BPA in the blood of uremia patients treated by dialysis has been conducted. Acidified blood and acidified SPE eluent were used to suppress the ionization of BPA and thus retain BPA on a C-18 column. BPA has two phenolic OH.
BPA, used to prepare polycarbonate and polysulfone materials used in the manufacture of medical devices and dental materials is an endocrine disrupter. Because polysulfone is biocompatible biomaterial it has recently been used to manufacture medical devices in contact with blood; polycarbonate and polysulfone are used in the manufacturing of the housing and the hollow fiber material, respectively of artificial dialyzers. Even if the amount of BPA leached from these components is low its endocrine-disrupting function will be hazardous to uremia patients.
Because they might be exposed for periods of more than 20 years (4 times per week, 4 h per each) so it must be concern how much BPA can migrate to body fluids of uremia patients during more than 20 years.
Polyacrylonitrile and poly(methyl methacrylate) are also regarded and used as biocompatible polymers therefore the migration of
the toxic and hazardous monomers of acrylonitrile and methyl methacrylate monomer must also be concerned. The author has previously described migration of toxic compounds from poly(methylmethacrylate) polycarbonate, and polysulfone used to manufacture the composite used dental material [3,9,16,17,34]. Although the selective determination of methyl methacrylate monomer from poly(methyl methacrylate) and the migration of the compounds from poly(methylmethacrylate) were described, migration of BPA from polycarbonate and polysulfone was not studied [17]. Organic solvent extraction of BPA from polycarbonate and polysulfone has also been described [16] but migration of BPA to the blood of uremia patient treated with an artificial dialyzer made of polycarbonate or polysulfone has not been studied so far. The author conducted the study to assess the risk to uremia patients of exposureto BPA during treatment by artificial dialysis.
Analysis of bisphenol A and SEP vs liquid-liquid extraction of bisphenol A and so on will be discussed later in detail.
Biocompatible polysulfone (PS) and polycarbonate (PC) are often used for the biomaterials of the medical device. Medical devices made of PS or PC must be sterilized before shipping. After sterilization toxic compounds can be produced as is MDA by γ-ray irradiation sterilization to PU. The degraded compounds after sterilization to polymers was identified, determined and clarified what compounds may inhibit bacterial growth [38]. An ethanol extract of the sterilized PS or PC was analyzed by liquid chromatography- mass spectrometry, UV photodiode array (210-350 nm) for identification and determination. Bisphenol A, 4,4’-dihydroxydiphenyl sulfone (bisphenol S) and 4-chloro-4'-hydroxydiphenyl were identified in PS sterilized with ozone gas. Bisphenol A was also identified in PC sterilized with ozone gas. Bisphenol A was not a major inhibitor of bacteria growth because the extent of inhibition did not correlate with the amount of bisphenol A produced. Inhibition by 4-chloro-4’-hydroxydiphenyl sulfone was greater than by bisphenol S and correlated with the amount produced; the major inhibitor might therefore be 4-chloro-4'-hydroxydiphenyl sulfone. The combined effect of bisphenol S and the 4-chloro-4'-hydroxydiphenyl sulfone might be a more realistic measure in the exact amount of inhibition. The order of cytotoxicity test was bisphenolA >4-chloro-4'-hydroxydiphenyl sulfone > bisphenol S, indicating bacteria inhibition test did not always identical to cytotoxicity test.
Several sorts of polymers are used for the fabrication of the medical devices. PS is synthesized from the 4,4'-dichlorodiphenyl sulfone and bisphenol A sodium salt[38]. PC is synthesized from the phosgene and bisphenol A [16,36].The toxicity of bisphenol A and bisphenol S has already been reported [39], however 4-chloro-4'-hydroxydiphenyl sulfone has not, so the toxicity of this compound must be clarified quantitatively.
Analysis of bisphenol A, bisphenol S and 4-chloro-4'-hydroxydiphenyl sulfone will be described later in detail.
Selective retention, removal and elution for analysis of toxic compounds to human health
Blood and saliva used are from the author. The saliva was sampled before breakfast from the author. Most of chemicals excepting compounds synthesized by the author were available in the market.
Pretreatment method described in the following text is applicable to analytical procedure as well as clinical treatment for human health. In this paper, solid phase microextraction (SPME), which is now quite popular among analytical chemist, was however omitted as the hardware of the equipment has still inferiority to be innovated and remained to be improved for attaining reproducible recovery rate.
As mentioned MDA is unstable upon heating, therefore HPLC was adopted for analysis combined with ECD detection. The eluent is a mixture of an aqueous solution (3 parts) containing 50 mM ammonium acetate for common ion effect as well as increasing ECD sensitivity and acetonitrile (l part) by volume ratio. This method was found to improve ECD detection sensitivity prevent MDA tailing and accelerate MDA elution[8,13]. An addition of ionic compound at around 50 mM is essential for ECD detection for attaining common ion effect and improving ECD detection. If no salt contained in the eluent, no successful ECD detection was done. ECD requires at least 50 mM salt in the eluent for electricity delivery in the eluent. ECD sensitivity increased with increasing applied voltage, but simultaneously decreasing selectivity, indicating less than 1000 mV will be most appropriate in terms of sensitivity and selectivity and prevent shortening cell life time [12]. Thus there are two reasons to add 50 mM to eluent.0ne is for common ion effect [8,13] and the other is to improve ECD detection sensitivity of aromatic amine. MDA was detected with ECD at 900 mV as well as UV at 250 nm. ECD detection was more selective and sensitive.When ammonium acetate in the eluent contained impurities, the base line by UV detection was fluctuate [12]. The detection limit of MDA by UV and ECD was 150 and 3 ppb (S/N=3 at the peak height) [12] respectively. ECD detection has inferiority that it can detect only for the compounds of functional group with relatively lower (mostly less than 1000 mV) oxidation- reduction potential. Greater voltage application may indicate greater sensitivity but lower selectivity in addition causing any damage to the electrode.
According to the development of column fabrication technology, residual silanol effect have been completely diminished by employing a silicone coating e.g. Capcel PakR from Siseido Co. (Tokyo Japan) or by employing high temperature for diminishing free silanol e.g. L-columnR from Chemical Inspection Co. (Tokyo) .The latter column due to its complicated fabrication technology no large size column is available as it is too expensive. These columns are completely free from residual silanol and heavy metal impurities in silica. The study of detection limit using these columns indicated at least 10 times lower (I mean more sensitive) than that by insufficiently endcapped C-18 columns due to narrower peak shape and higher peak height. This is due to no tailing as there exists no residual silanol [14,16] and even particle size of round silica at around 3 μm.
Liquid-liquid extraction procedures have been reported describing the repeated extraction with n-heptane-isoamyl alcohol (99:1 v/v) diethyl ether or benzene from alkalized serum in order to prevent MDA dissociation, however the reported recovery rate was unsatisfactory (from 70 to 80%) [40,42]. The author can admit this recovery rate even if it is lower as far as the recovery rate has reproducibility but it will not be certain for reproducible recovery rate can obtain or not.
In the author's preliminary procedure for liquid-liquid extraction one part of alkalized serum was extracted twice with seven parts of a mixture of chloroform and methanol at a ratio of 3/1 (v/v) and a satisfactory recovery of blood serum MDA was attained (98% in average, n=3) [11]. This procedure was often used for lipid extraction in biochemistry, but this procedure was troublesome, especially to carefully prevent trapping loss. Next the author innovated a more efficient method, in which only two to three parts of acetonitrile was added to one part of blood serum at a volume ratio for deproteinization and MDA extraction. This procedure required only single extraction and showed a satisfactory recovery of blood MDA (99%in average n=5) [8,10]. The reason why acetonitrile was thought to be satisfactory is because acetonitrile is an efficient deproteinization reagent as well as an efficient solvent of MDA. Most liquid-liquid extraction procedures for blood were required centrifugation after deproteinization, supernatant condensation of excessive organic solvent and the repeated extraction. The condensation by vacuum evaporation was undesirable due to MDA instability upon heating and loss of recovery [12,13] therefore SPE was studied as an alternative method.
In general SPE is easy to handle, to prepare for an automated system and requires less solvent consumption. Deproteinization, centrifugation, and condensation are usually unnecessary. It is more important to keep in mind that one cycle treatment with SPE will be almost identical to several tens of thousands of liquid-liquid extraction treatment, indicating one time of liquidliquid extraction is the same as the SPE with only one theoretical plate. The comparison of the eluent selection of several SPE columns was examined (Figure l) [8,9]. Eluent volume should be at least two to three times more than void volume of SPE column,otherwise no successful elution attained. For example, 100 mg of Bond ElutR C18 SPE from Varian Co.(Harbor City CA)has 120 μL of void volume therefore at least 240 to 360 μL will be required for successful elution.
In the elution of blood MDA, several sorts of SPE colurnns of Bond ElutR were tested (Table l) [8,9]. Reverse phase columns (columns except cation exchange in Table l)with a resin weight and void volume of 100 mg and 120 μL, respectively, were used. These columns were conditioned as shown in Figure 2. Conditioning rinsing and elution were carried out by a vacuum system [8,9].
C-1,C-2,C-8 and silica columns indicated lower MDA recovery rates whereas C-18 phenyl and cyclohexyl columns produced satisfactory recovery rates (Table l) [8,9]. The lower recovery by C-1 C-2,C-8 and silica columns and higher recovery by C-18 phenyl and cyclohexyl columns was obtained. The higher recovery by the latter can speculate that the Van der Waals binding and π-π interactions between benzene rings may play a major role for retention of MDA on SPE columns. On the contrary, binding of MDA to free silanol is thought to be minor from the result of lower recovery by silica column (Table l). This is thought to be due to water in serum interfering the combination of MDA to silanol. Recovery rate among C-1,C-2 and C-8 columns increased with increasing hydrophobicity of the columns (Table l), which was thought to be due to increase of Van der Waals binding capacity [8,9].
The cation exchange column produced a sufficient recovery (100% Table l) but requires a more complicated conditioning procedure than a reversed-phase columns. The inferiority is acidification of blood to charge MDA positively and centrifugation prior to SPE treatment to avoid protein [8,9].
MDA bound to the reversed phase column was not sufficiently eluted by methanol alone or HPLC eluent (a mixture of 50 mM ammonium acetate solution and acetonitrile at 3:1, v/v). However it was almost completely eluted by a strongly alkalized methanol solution (a mixture of methanol and l M ammonium hydroxide) (Figure l) [8,9]. The basic MDA bound to the reversed-phase column was normally speculated to elute more favorably after MDA was acidified to charge positively MDA. Based on this speculation the author initially carried out an elution experiment using an acidified methanol. However the results were opposite to the speculation, indicating the recovery was lower than alkalized methanol (recovery rate for methanol alone was 6.8% acidified was 10.2% and alkalized was 100% Figure l). The precise reason why the alkalized methanol was superior to the acidified methanol was not yet well clarified. The author’s speculation is as follows: alkalization will depress MDA and silanol dissociation and acidification will be opposite. From this of which amine charge in MDA or silanol charge will play more essential role for elution. According to the recovery result of the silica column (12% recovery Table l) binding of MDA to silanol is not significant. Acidified MDA promotes charging amine of MDA, but this does not result in favorable recovery (recovery rate of 10.2%). Silanol is depressed by acidification. Therefore, it will be concluded that both amine and silanol will not be essential factors to be considered.
The author finally considers the satisfactory result by alkalized methanol may be due to common ion effect. If so, elution mechanism is quite simple than ever been speculated however initial speculation method, which is opposite to common ion effect was so often reported for recommendable SPE eluents [40,42]. Further speculation is that acidification was insufficient to reduceblood pH due to blood buffer function at pH 7.4, positively charged MDA may partly bind to dissociated residual silanol in SPE column or positively charged MDA may not be sufficiently dissolved in hydrophobic eluent. If the last reason may be correct why favorable result by alkalized methanol was attained successful result can be clarified. This is because the undissociated MDA will be more favorably dissolved in hydrophobic eluent. Thus, the author considers two major reasons for favorable result using alkalized MDA. One is for favorable dissolution to the organic solvent (methanol) eluent of MDA and the other is for common ion effect. The speculation might not be correct, but the experimental result has a significant reproducibility to support the author's speculation. From this speculation, when determining optimum elution condition, we should simultaneously take into consideration the following factors: chemical charge situation in the eluent for the compound of interest, chemical and physical properties of the eluent and SPE column characteristics and the behavior of residual silanol. These indicate many factors must be simultaneously considered for favorable retention and elution.
MMA, DMPT and BPO analysis: The column for MMA, DMPT and BPO analysis was Capcell PakR C-18 SG-120 from Siseido Co. (Tokyo Japan). This column was completely endcapped with silicone coating. The eluent was a mixed solution of water and acetonitrile at a ratio of 4/1(v/v). The flow rate was l.2 mL/min. Detection was UV at 235 nm [14,17].
BA analysis: BA analysis was as follows: a Capcell PakR C-18 AG-120 column was used with an eluent of a mixture of acidified aqueous solution of water and acetonitrile at a ratio of 4/1 (v/v) adjusted to pH 3 with acetic acid. Acidification is for BA depression, otherwise BA will be eluted before void volume. Detection was by UV at 235 nm. The rest of the procedure was identical to MMA DMPT and BPO analysis.
Newly found toxic compound analysis: Methanol extract of dental material fabricated at room temperature of YunifastR from GC Co., Tokyo, was used for unidentified compound analysis. Using the gradient elution of HPLC shown later, many compounds including MMA, DMPT and BPO were eluted (Figure 4). Total peaks eluted were not completely identified, but identified compounds were shown as follows: aniline N-methyl p-toluidine p-toluidine BA 3-carboxy 4-N-methyl amino toluene 2-carboxy 4-N-methyl amino toluene 2-carboxy 4-amino toluene (3-amino 6- methyl benzoic acid) 3-carboxy 4-amino toluene (2-amino 5-methyl benzoic acid) 2-hydroxy DMPT 3-hydroxy DMPT 2,3-epoxy DMPT and o and p-N-methyl amino benzoic acid (Figure 4) [14,15]. In this paper hydroxy DMPT and epoxy DPMT was discussed in detail for reproducible determination.
The hydroxy derivatives of DMPT were synthesized by the author as follows: Each five gram of 2-amino-5 methyl-phenol or 3-amino-6-methyl phenol 10 gram of methyl iodide and 5 gram of potassium hydroxide were refluxed for 120 hours with stirring in 100 mL of anhydromethylethyl ketone. After cooling of the reaction mixture 300 mL of water was added for dissolution and neutralized, thereafter 300 mL of diethylether was added to extract the hydrophobic compounds. Ether layer was separated and evaporated. The residue after evaporation was distilled at vacuum condition. Thus 3-hydroxy-4-dimethylamino toluene and 2-hydroxy-4-dimethylamino toluene both of which are DMPT hydroxide compounds, were successfully prepared. No methoxy or benzene-methylated compounds were synthesized. In place of methyl iodide, when dimethylsulphonic acid was used for methylation reagent resulted in not only amino group, but also aromatic hydroxy group (phenolic OH) was also methylated as the methylation ability was so strong that methyl iodide was appropriate fort his purpose. 3-carboxy 4-N-methyl amino toluene and 2-carboxy 4-N-methyl amino toluene were prepared as follows: each five gram of 3-carboxy 4-amino toluene (2-amino 5-methylbenzoic acid) or 2-carboxy 4-amino toluene (3-amino 6-methylbenzoic acid) five gram of methyl iodide and five gram of potassium hydroxide were refluxed for 120 hours with stirring in 100 mL of anhydrous methylethylketone. Thus, 3-carboxy-4-N-dimethyl amino toluene, 2-carboxy-4-N-dimethyl amino toluene, 2-carboxy 4-N-methylaminotoluene and 3-carboxy 4-N-methylaminotoluene were prepared without production of methoxycompounds. After cooling of reaction mixtures, 300 mL of water were added for dissolution and neutralized thereafter the mixture was extracted with 300 mL of diethylether. The residue after evaporation was distilled at vacuum condition. Most of 3-carboxy-4-N-dimethyl amino toluene and 2-carboxy-4-N-dimethyl amino toluene were remained in aqueous layer and 2-carboxy 4-N-methylamino toluene and 3-carboxy 4-N-methylamino toluene were mostly extracted with diethytether.
The 3-carboxy-4-N-dimethyl amino toluene and 2-carboxy-4-Ndimethyl amino toluene were eluted earlier in Capcell PakR C-18 SG-120R HPLC than 2-carboxy 4-N-methylamino toluene and 3-carboxy 4-N-methylamino toluene. They were chromatographically separated and collected individually using collection scale C-18 column (50×250 mm linear velocity was identical to that of analytical column). MS fragmentation spectra and HPLC retention time of these synthesized compounds coincide with unknown compounds therefore the chemical structure of unidentified compounds can be successfully synthesized.
As these compounds have two opposite functional groups of aromatic amine and carboxyl (Figure 4) therefore appropriate and simultaneous SPE analytical conditions of these compounds are still under research.
The linear gradient elution was carried out using a mixture of 10 mM ammonium acetate/acetonitrile combined with HPLC column of Capcell PakR C-18 UG 120A (4,6×250mm). During 40 min ratio of 10 mM ammonium acetate/acetonitrile was changed from 9/l to l/9. In order to increase sensitivity by MS detection ammonium acetate was added to the eluent. The addition of ammonium acetate was not for common ion effect, but for increasing MS detection sensitivity. Flow rate was 1 mL/min, detection was by UV at 235 nm and 10 μL of methanol extract of YunifastR was injected to HPLC of HP 1050R from Hulett Packard Co. HPLC was connected with MS of TSQ 7000R from Finniganmat Co. at atmosphere pressure chemical ionization (APCI) mode. The information of mother ion molecular weight and MS fragmentation by HPLC-MS-MS mode was obtained and based on the information, chemical structure was successfully identified.
Identification of chemical structure of newly found toxic compounds: Reproducible separation of hydrophilic compounds was attained from BA to MMA using linear gradient elution. Using this separation method, chemical structure of unidentified compounds eluted from BA to MMA was identified. Using HPLC-MS at APCl mode, only information of mother molecular weight was obtained, therefore HPLC-MS-MS mode was essential to identify chemical structure from chemical fragmentation information. Inferiority of HPLC-MS at APCl mode was that hydrophobic compounds were not sufficiently detected however unidentified compounds in this case were DMPT derivatives therefore as they have aromatic amine in their chemical structure, so they were successfully detected.
Determination of unidentified compound in saliva: Each three sheet of 3×3×0.1 cm of YunifastR was immersed in 10 mL of saliva and the amount eluted to saliva was determined. Pretreatment of newly identified DMPT derivatives was carried out using SPE C-18 column with an identical manner to DMPT pretreatment procedure.
SPE procedure of MMA, DMPT and hydroxy DMPT in saliva: In saliva BPO was not existed as it is and immediately transformed to BA, therefore BPO in saliva was determined as BA. SPE treatment of MMA, DMPT and hydroxy DMPT was as follows: the C-18 column was conditioned with 2 mL of acetonitrile and 2 mL of 50 mM phosphate buffer at pH 7.5. Thereafter one mL of saliva was applied to the conditioned column vacuumed rinsed with 0.5 mL of 50 mM phosphate buffer at pH 7.5 and eluted with one mL of an alkalized acetonitrile with 50 mM phosphate buffer at pH 8. The eluent was trapped and 20 μL were applied to HPLC. Conditioning rinsing and elution were carried out by a vacuum system
SPE procedure of BA in saliva: BPO was stable in methanol, but when contacting with saliva or DMPT, BPO was immediately transformed to BA, therefore even though BA was not originally utilized in PMMA fabrication but BA was existed as a sort of artifact from BPO.
SPE procedure of BA in saliva was as follows: depression of ionization of BA was necessary to successfully retain in C-18 column. An aqueous solution at pH 3 (acetic acid) was added to the sample solution at a volume ration of 1/1 and mixed well prior to SPE application and carried out as follows: Bond ElutR C-18 column was conditioned with 2 mL of acetonitrile and 2 mL of acetic acid aqueous solution at pH 3. One ml of saliva was applied to the conditioned column. Saliva was two-fold diluted with an acetic acid aqueous solution at pH 3 prior to the conditioned column application. Thereafter, they were vacuumed, rinsed with 0.5 ml of acetic acid aqueous solution at pH 3 and eluted with one mL of acetonitrile acidified with acetic acid at pH 2.5. The eluent was trapped and 20 μL were applied to HPLC. Conditioning rinsing and elution were carried out by a vacuum system.
Liquid-liquid extraction was carried out by adding an identical volume of acetonitrile to serum for deproteinization and extraction of MMA and DMPT. The MMA peak showed an insufficient separation from serum admixtures in HPLC and insufficient recoveries of MMA and DMPT (84%and 62%for MMA and DMPT, respectively n=3) [9,17]. SPE treatment of blood MMA, DMPT and BPO was already mentioned in section of 2.2. 1. 6 [17]. The reason of the use of phosphate buffer at pH 7.5 was to depress ionization of DMPT, strong basic compound, but at pH 7.5 ionization of DMPT was incompletely depressed. How ever, due to characteristics of silica dissolution over pH 8, eluent pH over 8 was not applicable. As mentioned in 2.2.1.1, DMPT analysis was successfully attained without using phosphate buffer eluent. This is mostly due to the use of completely endcapped column of Capcell PakR to avoid residual silanol effect mentioned in advance. SPE of blood BA from BPO was also already described in 2.2.1 .7 [17].
Concerning the SPE eluent of BA acetonitrile, alkalized acetonitrile containing 50 mM sodium hydroxide or acidified acetonitrile adjusted to pH 2.5 with acetic acid were compared for eluting from SPE column. Acetonitrile alone showed an insufficient recovery (80%).Alkalized acetonitrile and acidified acetonitrile indicated 85% or 100% recovery, respectively, therefore acidified acetonitrile was superior to alkalized acetonitrile [17]. The successful reason using an acidified eluent for BA elution was because ion suppressed BA will be more favorably dissolved to the SPE eluent rather than retention on C-18, which is highly hydrophobic. Further speculation was that acidified solution was used during conditioning, so alkalinity may be suppressed due to acidified circumstances. As shown in citations of 9 and 17, no BA detection was in native blood. BPO in blood was also no detection in native blood and BA in place of BPO was successfully detected. BA came from BPO.
In SPE of blood MMA, DMPT and BPO, 50 mM phosphate buffer at pH 7.5 was used for column conditioning. The use of water or more than 50 mM phosphate buffer resulted in a lower recovery [17]. This is because an insufficient depression of DMPT ionization by water alone and excessive buffer ions at more than 50 mM may interfere with DMPT retention on the column. In DMPT elution, alkalized acetonitrile (phosphate buffer at pH 8) was more effective than acetonitrile or acidified acetonitrile due to the identical reason mentioned in SPE for MDA. These are favorable dissolution to the eluent than retention to reverse phase support [17]. Further speculation was that alkalized solution was used during conditioning, so acidity may be suppressed due to alkalized circumstances.
Acetonitrile also produced a satisfactory recovery for MMA (neutral compound) but not so much for DMPT (strong basic compound), which will be reasonably understood [17]. The favorable recovery reason for DMPT was due to identical reason for SPE elution of MDA (refer to 2.1.2). As MMA is a neutral compound, so it will not be effected by eluent pH. Satisfactory results have been produced, both when acidic acetonitrile was used for elution of acidic compound of BA and when alkalized acetonitrile was used for elution of basic compound of DMPT [17].
These results were different from already reported results for selection of SPE eluent however it is important that these experimental data have reproducibility thus it has a sound scientific rationale. Sound reason to be considered is common ion effect and favorable dissolution of the compounds of interest to the SPE eluent. This speculation may not be correct, however reproducible experimental results were obtained using the SPE eluent. The author considers that any experimental results with reproducibility will be much superior to only speculation without any experimental proof or computer simulation because speculation is speculation and simulation is simulation. Computer simulation cannot predict any interference peak elution with undefined retention time and it will not be useful when determining the compound of interest in complicated matrix such as body fluids.
Concerning SPE procedure, if compounds have carboxy group, they were treated with identical manner to BA treatment. If the compound of interest has no carboxyl group in the chemical structure identical SPE manner to MMA, DMPT and BPO will be utilized. When both carboxyl and amino groups exist in the chemical structure as is the case of newly identified compound further study will be required far appropriate SPE treatment.
Neutral compound of MMA and BPO was not affected to pH of eluent, so MMA and BPO elution were used identical procedure to DMPT treatment by SPE. The hydroxy DMPT has phenolic OH, but this acidity is weak, therefore this functional group was not effected so much for elution. Therefore, the SPE procedure of epoxy DMPT will be identical to that of original DMPT.
Result of newly identified compounds: The newly identified compounds of hydroxy DMPT were recognized to elute between BA and MMA elution. BA elution was confirmed by MH- (mother ion minus one proton). On the contrary, neutral compounds of MMA and BPO were not detected by MS (MH+ and MH-) at APCl mode due to less vaporization characteristic. The elution of newly found compounds of hydroxy DMPT was confirmed by MS (MH+), HPLC with UV detection and by coincidence of elution time with standard compounds synthesized.
Unidentified hydrophilic compounds were determined their chemical structure as hydroxy derivative of DMPT, 2,3 epoxy DMPT and carboxyl derivatives of DMPT from their molecular weight and MS fragmentation pattern as well as coincidence of elution time of standard compounds. Their retention time was 6.7 min, 10 min and 11.5 min in this order. The retention time of BA, MMA and DMPT was 4.3 min, 20.3 min and 27 min, respectively. The elution of N-methyl-p-toluidine was confirmed to elute just after MMA elution. The retention time of this compound was 20.7 min and that of MMA was 20.3 min. The elution of this compound was confirmed from MS fragmentation. These compounds were confirmed from methanol extract of YunifastR as well as the mixed solution of DMPT and BPO. This possibility indicates that unidentified compounds may be produced from the reaction of DMPT and BPO during PMMA fabrication. This compound was also treated with identical SPE manner to DMPT treatment of SPE.
The 2,3 epoxy DMPT was stable in methanol solution but when contacted with saliva the epoxy compound changed immediately to 2 or 3 hydroxy DMPT. It was recognized that BPO was immediately converted to BA when BPO was contacted with saliva or DMPT. As 2,3-epoxy DMPT and BPO were highly reactive therefore these compounds may be highly toxic. The degree of toxicity was not always parallel to the eluted amount. Serum extraction of MMA, DMPT and BA from YunifastR was 32.04 μg/g, 66.44 μg/g and 2.3 μg/g and the cytotoxicity data of lC50 (μg/ mL) of MMA, DMPT, BA and BPO using Balb 3T3 cell was 4400, 1500 28.7 and 22 [17]. It will be problematic when considering that epoxide compound may indicate the greatest toxic but as this compound will be transformed to hydroxy compound immediately when contacting with saliva or blood, therefore the cytotoxicity test of epoxide compound was not successfully attained until now.
The hydroxy DMPT compounds found in saliva were the total of originally existed in saliva plus those from 2,3 epoxy DMPT, but the differentiation of origin of these compounds in saliva was extremely difficult and the effort for differentiation was meaning1ess. The eluted amount of 2 hydroxy DMPT and 3 hydroxy DMPT into saliva in successive three days was 10.7 μg/g and 15.8 μg/g (n=3) respectively [17]. The amount of epoxy DMPT in saliva was not attained due to transformation to hydroxy DMPT immediately when contacting with saliva. The elution of successive three days will be minimum due to putrefaction of saliva for further period of immersion thus real elution amount will be much greater due to much longer period contact with dental material with saliva or blood through teeth.
The BPO in saliva was determined as BA at 3.5μg/g.
Author's blood was sampled for blood urea analysis. Native blood and denatured blood with acid were centrifuged for ultrafiltration at 13,000 rpm (10,000 g) for 40 to 60 minutes and the supernatant was applied to pretreatment method. Ultrafiltrator used was Kokusan Co. H-1300R in Tokyo. Membrane of ultrafiltration is CentrifreeR from Amicon Co. made of cellulose with cut-off molecular weight of 5,000 daltons. Native blood and denatured blood are for analysis of free and total urea, respectively.
For pretreatment method, an automated SPE and a dialysis were compared. SPE of blood was as follows: supernatant after ultrafiltration was applied to the conventional strong cation exchange column of Bond ElutRSCX (500 mg of resin weight and 0.6 mL of void volume). The SCX column was conditioned with 3 mL of methanol followed by 3 mL of water at a flow rate of 3 mL/ min. One mL of blood was applied to the conditioned SCX column at a flow rate of 0.3 mL/min and rinsed with l mL of water at a flow rate of 3 mL/min. The retained urea was eluted with 4 mL of 5%phosphoric acid at the flow rate of l mL/min. These procedures were carried out using the automated SPE equipment of BenchMateR from Zymark Co. controlled with computer [43]. Automated SPE was significantly superior to manual type SPE in terms of flow rate control, which will significantly affect to variation of recovery rate.
Automated dialysis was carried out as follows: The ASTEDR and the trace enrichment column (TECR) from Gilson Co. were used. A polymer-based strong cation-exchange resin column was used for TECR, which served for condensing dialysate. The resin weight was 20 mg. The column was conditioned with l.5 mL of l M sulfuric acid followed by 0.9 mL of water at a flow rate of 2 mL/min. The other conditions were as follows: dilutor l, 0.01% Tritonx 100; dilutor 2, 5 mM phosphate buffer(pH 7.4). The cutoff molecular weight for dialysis membrane made of cellulose was 15,000 daltons [43].
Urea analysis by conventional strong cation exchange resin column was carried out as follows: After pretreatment, sample was applied to the conventional strong cation exchange resin column of MCI GEL CK 08SR from Toso Co. 4.6× 150 mm, 11-14 μm particle diameter. Other conditions were as follows: eluent, l mM HCl solution; flow rate l mL/min; detection, 200 nm, application volume,20μL;column temperature, 35°C. HPLC equipment and UV detector used were PU-980R and PU-970R, respectively, from Nihonbunko Co. Tokyo.
The comparison of separation efficiency between ion chromatography use column with smaller capacity and conventional strong cation exchange resin column with greater capacity was studied and the result will be described later [44].
MECC analytical condition was as follows: running buffer, 75 mM sodium dodecyl sulfate (SDS) 10 mM hydrogenphosphate 6 mM tetraborate pH 9.2; applied voltage: 25 kv current: 70 μA; effective capillary length:68 cm, inner diameter 75 μm. MECC equipment is CAPI-3100R with photodiodearray detector from Otsuka Electronics Co.(Osaka, Japan).
As other pretreatment method, supercritical fluid extraction (SFE) will be available [45]. However, this method has a restriction only applicable to the vaporizable hydrophobic compound. As urea is hydrophilic, so current SFE technique is not effective for urea extraction. If this restriction will be conquered in future, this method will be more appropriate because extract is liquid gas, therefore it is unnecessary for evaporation and condensation.
Blood is a complicated matrix indicating that blood contains many compounds to interfere urea analysis. In order to attain satisfactory separation with sufficient resolution free from blood admixtures, blood urea must be pretreated. The recovery of blood urea from SPE cation exchange column was almost 100%. No or quite low recovery of blood urea was attained when using reversedphase column such as C-18 [20,22]. This means urea did not retain sufficiently on the reverse phase columns because urea was highly hydrophilic compound. In case of C-18 column urea was eluted at around void volume without separation from blood admixtures. That's why the author used complicated column switching method combined with immobilized urease column for post column Indophenol colorimetry [22] or immobilized urease precolumn method [21] combined with complicated column switching procedures. Both methods indicated satisfactory separation from blood admixtures, however it has originally inferiority that the methods were not easily applicable to routine analysis due to its complexity, which was a weak point of the methods [21,22]. At that time when the papers were published, column switching was manually conducted by calculating the switching time. In recent an automated column switching equipment is commercially available, therefore the author's previous set-up manual methods will be more easily applicable for routine analysis of clinical chemistry using automated column switching procedures.
SPE procedure for blood urea was carried out by acidified condition combined with SCX column. In order to elute urea successfully, stronger acidity eluent will be appropriate in terms of successful elution and transformation to H type of SCX column. In SPE treatment of blood urea pretreatment mechanism and column differed from MDA, BA and DMPT used. Pretreatment mechanism and column used for MDA, BA and DMPT procedure were partition mechanism and reverse phase columns. Those for urea are ion exchange mechanism and cation exchange column. This is because acidic eluent was successful for weakly basic compound of urea. As mentioned in advance urea can't be successfully analyzed with reverse phase columns because urea did not retain on them successfully [20,22].
Differential analysis of free from bound urea can be attained by ultrafiltration. For differential analysis of bound from free urea, ultrafiltration with centrifugation is one method. The alternative method is dialysis.
Ultrafiltration can be replaceable to dialysis. Using automated dialysis of ASTEDR dialyzate was condensed on the condensed column (TECR enrichment column). The compounds accumulated on the condensed column (TECR column, strong cation exchange column with Na type) were eluted with 1 mM HCl of HPLC eluent, which was identical to SPE treatment based on same reason [43]. HPLC chromatograms after strong cation exchange automated SPE and the automated dialysis were presented in citation 43. Urea was determined using standard addition method [43].
When compared chromatograms SPE and dialysis treatments[43] blood urea was separated with baseline separation from blood admixtures in both cases [43]. If mentioning in detail, in the chromatogram after SPE treatment only urea was eluted with sufficient separation from other blood admixtures indicating no compounds interfered with blood urea analysis. However, in case
of dialysis [43] urea can be separated anyhow with baseline separation from blood admixtures but separation after SPE was significantly superior to that after dialysis [43]. When considering blood urea analysis with SCX column capacity, urea amount contained must simultaneously be considered to attain successful baseline separation.
Inferior point of dialysis was a lower recovery of blood urea at around 10% mostly depending on TECR capacity rather than dialysis system, so by improving TECR capacity this problem will be resolved. However it will remain the problem as follows: if TECR capacity may differ, separation efficiency may change and peak broadening may differ thus separation efficiency of chromatogram after dialysis will be required to be improved because even though chromatogram after dialysis treatment indicated sufficient base line separation from admixtures but an insufficient separation of urea from blood admixtures may occur if TECR capacity may change [43]. Reverse elution is an alternative approach to resolve successfully without diminishing separation efficiency.
Automated dialysis and automated SPE equipments ASTEDR and ASTECR from Gilson Co. respectively can be connectable to HPLC for on-line analysis. Additionally if an autosampler will be installed to them the automated pretreatment system can be attained successfully. The hyphenated and automated set-up system with autosampler-automated SPE or automated dialysis and HPLC in combination will currently be available in the market.
For the author additionally requires to attach the on-line autocentrifugation (ultrafiltration) system for differentiation of free from bound type compounds in blood. However this kind of equipment is not available in the current market. This hyphenated technique for differential analysis will be desirable for routine clinical test to accurately diagnosis.
Simultaneous uremic toxins analysis of urea, uric acid, creatinine and methylguanidine was carried out with reverse phase column combined with complicated column switching method and online immobiiized urease column for post column method for urea analysis [19,22]. Among these uremic toxins only urea did not significantly retain on the reversed-phase HPLC columns [22]. Pretreatment method of blood urea with sufficient recovery has not been reported so far so the author considered the appropriate pretreatment method of blood urea.
In the preliminary experiment, the author carried out the experiment using an ion chromatography column with a smaller ion exchange capacity, however the separation of blood urea from blood admixtures was not satisfactory [44]. For the alternative method, the author considered the use of the conventional strong cation exchange column (H type)with a greater ion exchange capacity for the differential analysis of blood urea from endogenous ammonium[44]. Endogenous blood urea eluted faster than endogenous ammonium when using strong cation exchange column. Urea was detected at 200-210 nm [44]. By using this procedure a simpler procedure for differential analysis of urea and endogenous ammonium can be attained. Urea can be successfully separated from blood admixtures using the conventional strong cation exchange column (H type) [44].
As mentioned in 2.3.9 separation of endogenous blood urea from endogenous ammonium and admixtures can be successfully attained using a conventional strong cation exchange resin column. The problem unresolved yet was that the retention time of urea was unstable even though completely conditioned with strong acid to H type for suitably prolonged period [44]. Retention time of urea gradually increased (retention time keeps greater, however, not always constantly), speculating gradually changing H type in the column. This will be due to SO3H functional group in the interior of pore of silica support. If any deterioration to the column with blood may occur, shorter elution will be observed due to a smaller ion exchange capacity by clogging of protein to the column. As mentioned in advance, speculated reason of gradual increase of retention time may be due to a gradual change to H type of the interior functional group (SO3Na→SO3H) in silica pore.
This problem was observed in both cases in MCIR gel from Mitsubishi Co. and TSKR gel from Toso Co. This phenomenon was not well reasonably clarified and explained yet. Therefore, if the reader will determine from the peak height of urea, this phenomenon may cause a trouble to attain reliable data. When determining from peak area using computer calculation, this problem will be somewhat diminished but still remains the problem how to set baseline of urea peak. Depending on baseline setting, peak area will be significantly differed, especially if the peak indicates significant tailing which is so often observed in the ion exchange column chromatography for basic compound analysis.
Up to here urea analysis using HPLC has been described. In the next urea analysis using capillary electrophoresis (CE) will be described.
In MECC using SDS as a micellar compound over critical concentration blood urea migrated at around void volume overlapped with blood admixtures by direct blood injection, indicating undesirable separation, therefore other mode such as capillary zone electrophoresis (CZE) will be required [3]. As being mentioned in advance single urea analysis in any matrix urea analysis will be attained by CZE, but simultaneous analysis of urea and hydrophobic compounds such as uric acid will be required by MECC mode. MECC mode is identical to reverse phase mode therefore failure of urea separation by MECC is identical to HPLC with C-18 column.
Furthermore, blood uric acid analysis by MECC indicated insufficient separation of uric acid peak from blood admixtures, mostly blood proteins [3], indicating inferior to the reversed-phase HPLC (C-18 column) [3,19-22]. This result was identical to the published paper [23]. HPLC can separate blood compounds successfully by changing sort of columns, so in terms of blood urea analysis, HPLC was thought to be superior to MECC for selective blood urea and uric acid analysis.
Concerning blood urea analysis, HPLC is thought to be superior to CE in terms of appropriate selection from several sorts of separation mechanisms. However, CE technology is advancing day by day, so this status is not always unchanged. In case of only blood urea analysis not for the simultaneous analysis of blood urea and uric acid, CZE mode is thought to be more appropriate[3]. CZE will be superior to conventional isotachophoresis in terms of ion analysis.
Inferior points of CE is that capillary column utilized is only bare or coated silica column with or without addition of critical concentration of micelle compound for MECC for the former indicating less selection of separation mechanisms compared with separation mechanisms applicable to HPLC [3].
Separation time by CE was much shortened due to a greater theoretical plate number. Sample volume of CE was much less than the conventional HPLC excepting capillary HPLC [3]. This will be desirable for clinical analysis because tiny sample volume is favorable to patients. The most inferior point of CE to be improved is less reproducibility of injection volume migration time, peak height and other factors affecting to the accurate determination. Less reproducible data will not be well evaluated. When this inferiority will be conquered by improvement with innovated and advanced technology CE will become desirable analytical equipment in clinical analysis.
The author tried to find out an appropriate method of pretreatment to present a sufficient and reproducible recovery result [26]. To human serum samples of between 5 and 8 mL in which PA MEHP and DEHP were added 20 μL of acetonitrile-water (60:40) solutions of PA, MEHP and DEHP (10.0, 39.4 and 39.08 mg/mL), shaken well, allowed to stand overnight at 4°C and used for the study of the deproteinization as described below.
At first, the effect of deproteinizing eluate on the elution of DEHP from serum was studied. Addition of l M sodium hydroxide with acetonitrile to serum for deproteinization resulted in nearly 100% recovery of DEHP [26]. The possibility of DEHP being hydrolysed with sodium hydroxide was discounted. Deproteinization with acetonitrile alone did not give a satisfactory recovery of DEHP, and an acid added in place of l M sodium hydroxide also gave a low recovery [26].
The author then studied the effect of deproteinizing eluents on the elution of MEHP in serum. Although the same pretreatment of serum as with DEHP resulted in about 90% recovery [26]. a variety of attempts were made for a higher recovery. To suppress ionization of MEHP, phosphoric acid was added to serum and further a mixture of diethyl ether-methanol (2:1) was added followed by stirring centrifugation and injection of the diethyl ether layer of the supernatant into the HPLC system. The immediate recovery obtained was about 86% but a recovery of 100%was attained when the mixture was allowed to stand overnight at 4 oC after stirring without centrifugation. The mixture was stirred centrifuged and the diethyl ether layer of the supernatant injected into the HPLC system. Recovery of MEHP and DEHP in human plasma was also about 100% using the above deproteinization method. Inferior points are the use of different solvent for DEHP and MEHP [26]. This is in general inferior point of liquid- liquid extraction. In addition it is inferior that no satisfactory recovery method of PA was attained with liquid-liquid extraction [26], therefore the author conducted the ultrafiltration method described below.
With the same materials as used for deproteinization, ultrafiltration was studied as a pretreatment method [26] Of the PA, MEHP and DEHP in human serum about 77% 3%and 0 % respectively were found to be present in the ultrafiltrate [26].Addition of acid to the serum increased the amount of PA in the ultrafiltrate to about 80% but it caused little change in amounts of MEHP and DEHP. An increased amount of acid did not alter the amounts of PA, MEHP and DEHP in the ultrafiltrate, but dilution of serum with water increased the recovery; in particular PA was found for the most part in the ultrafiltrate after addition of acid to a fourfold dilution of serum with water. PA in human plasma was about 100% recovered by this ultrafiltration method [26].
To l mL of human serum as used for deproteinization 3 mL of water and 0.1 mL of 50% phosphoric acid were added and shaken vigorously. Thereafter l mL of the mixture was applied to Sep-PakR C-18 column and eluted with 8 mL of methanol or acetonitrile.
The solvent was evaporated and the residue was dissolved again in l mL of the eluent for analysis of DEHP MEHP and PA. The recovery rates of them were satisfactory.
No difference was found in the recovery ratio between acetonitrile and methanol. Experiments under various conditions revealed that the Sep-PakR C-18 column method for the recovery of DEHP MEHP and PA in human serum was quite superior to the methods described in 2.4.1 and 2.4.2 in terms of the satisfactory recovery rate, easy handling sufficient reproducibility less consumption of organic solvent and so on.
Blood of uremia patients (ca 10 mL) was sampled before dialysis and after 4 h of dialysis treatment to confirm whether or not BPA in the blood was originating from the dialyzer. After addition of heparin blood was centrifuged at 5000 rpm for 20 min. The supernatant plasma thus obtained was stored under refrigeration at 4°C; under these conditions. It was stable for two weeks.
Before analysis the plasma sample was treated by automated SPE on l mL Varian BondElutR cartridges containing 120 μL (100 mg) C-18 resin. The endcapped C-18 SPE cartridge was conditioned with methanol then water (3 mL of each) at a flow rate of 3 mL/ min. One mL of plasma from the uremia patient was applied to conditioned cartridge at 0.5 mL/min and the cartridge was rinsed with water (l mL) at l mL/min. BPA was then eluted with 5% phosphoric acid (4 mL) at 0.5 mL/min [37].
Conditioning rinsing and elution during SPE were performed under vacuum by means of automated Varian BenchMateR equipment, otherwise reproducible recovery could not be achieved successfully.
HPLC was performed with a Hewlett-Packard HP 1050R chromatograph equipped with UV detection (Shimadzu Kyoto Japan SPD 2AR) and ECD (Yanagimoto Tokyo Japan VMD-501R). BPA in the blood of uremia patients was determined by means of ion-suppression chromatography on a 250 mm x 4.6 mm i.d.endcapped Capcelpak C-18- UG 120AR column. Isocratic elution was performed with 3:1 (v/v) 10 mM ammonium acetateacetonitrile as the eluent, at a flow rate was l ml/min; under these conditions the retention time of BPA was 7.6.min [37]. The injection volume was 10 μL.
Because BPA has two phenolic hydroxy groups in its chemical structure, it can be detected both by UV absorption (at 235 nm) and by ECD at 900 mV as is MDA. The order of connection of the detectors at the column outlet should be UV then ECD -if the ECD is connected between the column and the UV detector the ECD cell will be damaged by back pressure from the UV detector. HPLC-MS was performed by connecting the chromatograph to a Finnigan MAT TSQ 7000R with APCI. The mass range scanned was between 65 and 800 daltons. BPA was identified by comparison of its UV and MS spectra with those of standard BPA.
The limit of detection for a signal-to-noise ratio (S/N) of 2 was 0.002ng/mL; because accuracy and precision were both within20% this can be regarded as an approved detection limit. The limit of determination of BPA by ECD, using the above procedure, was 0.02 ng/mL (0.02 ppb) in plasma. Use of automated SPE with the BenchMateR equipment resulted in 99.2 ± 2.77%(average ± SD; n=7) recovery of BPA from blood plasma. The response to BPA was linearly dependent on concentration within the range 0 to 80 ng/mL: the correlation coefficient was >0.99.
When SPE was performed manually the recovery varied significantly and was less than that obtained by use of automated SPE. The reproducibility of manual SPE was significantly inferior to that of automated SPE because accurate pressure control is difficult in the former. Recovery data obtained by manual SPE were thus unreliable with significant variation of the result.
SPE vs liquid-liquid extraction and the formation of artifact with solvent extraction
Liquid-liquid extraction was a conventional pretreatment method for isolation and purification of the compound of interest in complicated matrix. The inferior points of this method were copious consumption of organic solvent for extraction requiring further condensation, which may result in loss of recovery during evaporation condensation time consuming due to repeated extraction or artifact formation of interest compound with extraction solvent [26,35,45,46].
An artifact formation is a problem associated with liquid-liquid extraction. An example of artifact formation was as follows: in case of amine compound extraction with methanol or ethylacetate formaldehyde from methanol causes Mannich reaction with methylol linkage to produces amine oligomer artifact for the former and acetylated of the compound of interest for the latter. These artifact compounds indicate more carcinogenicity [15-17]. In addition, they produce lower recovery rate of compound of interest. Artifact formation for example, can be prevented by replacing extraction solvent of methanol with ethanol.
During a vacuum evaporation/condensation process compound of interest was so often vaporized without being successfully trapped and may cause a reduction of recovery and thermal decomposition [35,45,46]. Liquid-liquid extraction required a greater amount of consumption of organic solvents, which was hazardous to chemists. The recovery rate of single treatment of liquidliquid extraction was in general less recovery than SPE. This is because a single treatment of liquid-liquid extraction was almost identical to SPE column with one theoretical plate. In general, SPE did not require condensation and could be condensed using amount of eluent amount almost identical to column volume.
As mentioned, when ethyl acetate was used as an extraction, compounds with hydroxy or an amino group was acetylated, which causes a reduction of recovery rate. Artifact of acetylated compound was generally more mutagenic and toxic, thus this may lead to misunderstanding to the researchers that they may consider to extract strongly toxic compounds and of course this will cause lower recovery rate. Analyticai chemists should keep in mind well about artifact formation during solvent extraction by reacting solvent with the compound of interest, otherwise they may have misunderstanding to extract stronger toxic compounds in their experiment. In that meaning almost 100% recovery rate is most desirable to avoid misunderstanding, otherwise less recovery may indicate a possibility of artifact formation during extraction.
In order to avoid artifact formation, one approach is to replace to appropriate extraction solvent. The other is to use SPE in place of liquid-liquid extraction. Inferiority of SPE is that if SPE procedure is carried out manually, vacuum pressure control was very difficult, which results in less reproducibility of recovery rate. In that meaning, automated SPE such as BenchMateR, ASPEC or RapidTraceR will be recommendable because these SPE equipment are controlled by computer thus pressure control reproducibility is much superior to manually controlled SPE. Among them Rapid TraceR from Varian Co. will be most recommendable because it is cheapest and handy type with 10 samples treatment automatically in successive run.
Conclusions
Artifact formation during solvent extraction including SPE is problematic but this phenomenon was so often overlooked [47]. The conventional liquid-liquid extraction has inferiority compared with SPE in several reasons mentioned in advance.
Eluent of reverse phase columns for basic or acidic compounds was different from conventionally reported results for SPE elution. Suitable experimental result of eluent constitute was opposite from the initially speculated eluent. It is more important to recognize that the experimental result has reproducibility. Reproducible result is essential. Explanation of the experimental result was most probably due to common ion effect, favorable dissolution to the eluent and other speculated reasons.
Newly identified toxic artifacts were produced during PMMA fabrication. These were successfully identified using HPLC-MS-MS at APCl mode. These were derivatives of starting compounds. During reaction of DMPT and BPO most of newly identified compounds were produced. Epoxy derivative compound of DMPT was further converted to other compounds when contacting with body fluids such as blood or saliva. Some artifact derivative compounds are toxic and they have both aromatic amine and carboxyl functional groups in their chemical structure therefore appropriate SPE procedure must be further studied.
Eluent of reverse phase columns for basic or acidic compounds was different from reported results and also the author's initial speculation. Most important fact is to recognize that the experimental result has a satisfactory reproducibility or not. The reproducible experimental results indicate any truth the researchers may overlook. Explanation of the author's experimental result was mainly common ion effect and favorable dissolution to the eluent. As the experimental result has a reproducibility with satisfactory recovery so the experiment has a meaningful scientific rationale.
References
- Shintani H (1995) Formation and elution of toxic compounds from sterilized medical Products methylenedianiline formation in polyurethane ,J..Biomater. Appl.10, 23.
- Shintani H (1992) Gamma-radiation and autoclave sterilization of thermoplastic and thermosetting polyurethane, J..Radiation. Steril. 1, 11 .
- Shintani H (1998) Adsorption and its applications in industry and environmental protection A Dabrowski (Ed) Elsevier New York,77.
- Shintani H (1996) Formation and elution of toxic compounds from sterilized medical products: toxic compound formation from products Radiat. Phys.Chem 47,139.
- Shintani H (1991) Polym.Degradation, Stabil., 32,17.
- Shintani H, Hirata N (1995) Gamma-ray irradiation, autoclave and ethylene oxide sterilization to thermosetting polyurethane: Sterilization to polyurethane. Radiat. Phys. Chem. 46, 377.
- Shintani H (1995) “The relative safety of gamma-ray, autoclave and ethylene oxide in dialyzer rinsing fluids:thermosetting polyurethane”.Biomed.lnstrument. Technol., 29, 513.
- Shintani H (1991) Solid-phase extraction of a carcinogen, 4,4'-methylenedianiline, inm serum.J.Anal.Toxicol.15 198.
- Shintani H(1992) Solid-phase extraction (SPE) and HPLC analysis of toxic compounds and comparison of SPE and liquid-liquid extraction. I. Analysis of 4,4/-methylenedianiline in serum. II. Analysis of the compounds of dental materials, J.Liq.Chromatogr.,15, 1315.
- Shintani H (1992) Solid-Phase Extraction and High-Performance Liquid from – irradiated Polyurethane J.Chromatogr.,600, 93.
- Shintani H and Nakamura A(1989) Analysis of the carcinogen 4,4 methylenedianiline (MDA) in gamma-ray and in autoclavesterilized polyurethane- Frezenius Z. Anal. Chem. 333,637.
- H and Nakamura A (1989) Analysis of a carcinogen, 4,4'-methylenedianiline, from thermosetting polyurethane during sterilization. J.Anal. Toxicol. 13, 354.
- Shintani H and Nakamura A(1991) Formation of 4,4′-methylenedianiline in Polyurethane potting materials by either γ-ray or autoclave sterilization.,J. Biomed.Mater.Res., 25,1275.
- Shintani H (1995) Japan.J.Medical. lnstrumentation, 65,249.
- Shintani H (1994) Japan.J.Medical. lnstrumentation, 64,345.
- Shintani H (1995) J.Liquid Chromatogr.. Clin. Anal., 18,613.
- Shintani H, Tsuchiya T and Nakamura A (1993) Solid phase extraction and HPLC analysis of toxic components eluted from methyl methacrylate dental materials J.Anal.Toxicol.17,73.
- Shintani H(1995) Japan.J. Medical. Instrumentation, 65,486.
- Shintani H, Wojcik A.B Tawa R and Uchiyama S (1994) Analytical Applications of lmmobilized Enzyme Reactors.S. Lam and G. Malikin (Eds) Blackie Academic & Professional, Glasgow 131.
- Shintani H and Suzuki H(1991)Bioinstrumentation and Biosensors, D.L Wise (Ed),Marcel Dekker, New York, 181.
- Shintani H and Ube S (1985) J. Chromatogr. 344 145.
- Shintani H (1986) Simultanious detemation of serum uremic toxins, cations and anition and urea determination by post-column colorimetry using immobilized anzyme., J.Chromatogr.,378,95.
- Schmutz A, Thormann W (1994) Determination of pharmaceuticals in plasma by capillary electrophoresis without sample pretreatment reproducibility, limit of quantitation and limit of detection., Electrophoresis 15,51.
- Shintani H (1996) Handbook of Capillary Electrophoresis Application H.Shintani and J. Polonsky (Eds) Blackie Academic & Professional London,499.
- Fujiwara S , Todoroki H Ohhashi H, Toda J and Terasaki, M (1990) Application of an Acid Urease to Wine: Determination of Trace Urea in Wine.,J.Food.Sci.,55,1018.
- Shintani, H (1985) J.Chromatogr., 337,279.
- Shintani H(2000) Chromatographia,52,721.
- Woodward K.N(Ed) (1988)Phthalate esters:Toxicity and metabolism, CRC Press.
- Vessman J. and Rietz G.(1974) Determination of di(ethylhexyl) phthalate in human plasma and plasma proteins by electron capture gas chromatography.,J.Chromatogr., 100 153.
- Rock G ,Secours V.E ,Franklin C.A , Chu I and Villeneuve D.C(1978)Fed. Proc.,27,442.
- Draviam E.J , Kerkay J and Pearson K.H (1980) Separation and quantitation of urinary phthalates by HPLC., Anal.Lett. 13 1137.
- Shintani H (1984 ) Bunseki Kagaku (Analytical Chemistry in Japanese) 33,314.
- Robinson J.L, John,J , Safa, A.L, Kirkes K.A, Griffith P.E(1987) Evaluation of disposable cartridges for trace enrichment from aqueous solutions.,J.Chromatogr.,402,201.
- Shintani H (1998) J. Liquid Chromatogr. Clin.Anal.,21 2297.
- Shintani H (1995) J.Liquid Chromatogr., .Clin.Anal. 18,2167.
- Shintani H (1996) Comparison of solid-phase extraction and dialysis on pretreatment efficiency of blood urea analysis.,J.Chromatogr.Sci.,34,92.
- Shintani,H (2001) Chromatographia,53,331.
- Shintani,H (2003) Chromatographia,58 193.
- Katayama M Matsuda Y Sasaki T Shimokawa K Kaneko S.et al (2001) Biomed.Chromatogra. 15,437.
- Tortoreto M. Catalani P. Bianchi M Blonda C Pantarotto C et al(1983) Determination of 4,4'-diaminodiphenylmethane in blood by gas-liquid chromatography with electroncapture detection J.Chromatogr.,262,367.
- Cocker J Brown L.C. Vvilson H.K , Rollins K (1988) A GC/MS method for the determination of 4,4'-diaminodiphenylmethane and substituted analogues in urine, J.Anal.Toxicol. 12, 9.
- Bowman M.C (1978) J.Assoc.Off.Anal.Chem.,61 1253.
- Shintani H (1996) Comparison of solid-phase extraction and dialysis on pretreatment efficiencyof blood urea analysis.,J.Chromatogr.Sci.,34,92.
- Shintani H (1994) Selection of columns for analysis of blood urea.,J.Liq. Chromatogr.,17,1737.
- Shintani H , Inoue G (1994) Determination of blood urea using cation- exchange column solid-phaseextraction combined with hplc .,Bunseki Kagaku,43, 805.
- Shintani H (1996) Japan.J. Medical Instrumentation,66,414.
- Shintani H (1994) Nippon no Shouhisha to Ryuudousei Seiyaku (Japanese Consumers and Liquidity Constraints: A Test Based on Credit Information).Osaka Economic Papers 44, 41-56.





