Prokaryotic Expression of VP7 Gene and Subunit Vaccine Preparation of Bovine Rotavirus
Wei S1,2, Che T3, Song C2, Tian F2, Ma Z1,*
1 The Key Bio-Engineering and Technology Laboratory of National Nationality Commission, Northwest University for Nationalities, Lanzhou, China.
2 Life Science and Engineering College, Northwest University for Nationalities, Lanzhou, China.
3 Lanzhou Baiyuan Company for Gene Technology, Lanzhou, China.
*Corresponding Author
Ma Zhongren,
The Key Bio-Engineering and Technology Laboratory of National Nationality Commission,
Northwest University for Nationalities, Lanzhou,
China.
Tel: +0086-931-2938310
E-Mail: mazr_minhai@163.com
Article Type: Review Article
Recieved: January 17, 2015; Accepted: April 20, 2015; Published: April 22, 2015
Citation: Ma Z, et al., (2015) Prokaryotic Expression of VP7 Gene and Subunit Vaccine Preparation of Bovine Rotavirus. Int J Vaccines Res. 02(1), 1-6.
Copyright: Ma Z© 2015. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
Abstract
The aims was to construct a prokaryotic expression plasmid and develop a subunit vaccine for bovine rotavirus (BRV), also to investigate the efficacy of this vaccine. BRV total RNA was extracted from M-A104 cell infected by the strain GSB01 of bovine rotavirus. Total RNA as template, BRV VP7 gene was amplified with real time fluorescence quantitative PCR (qPCR), PCR product was cloned into pEASY-T3 vector. The pEASY-T3-VP7 plasmid was recombined into the prokaryotic expression vector pET32a (+) after it was digested using double enzymes. The pET32a-VP7 and VP7-LTB used for prokaryotic expression were constructed, which were converted into Escherichia coli BL21 (DE3) competent cells. After IPTG induction and SDS-PAGE analysis were performed, the fusion proteins of pEASY-T3-VP7 and VP7-LTB with molecular weights of 42.2kD and 53.2kD were acquired respectively. Such a gene engineering bacteria for expressing VP7 protein within BL21(DE3) cells was constructed successfully. The mice immunized using both fusion proteins of pEASYT3- VP7 and VP7-LTB could promote VP7 IgG antibody production (8.33 vs 17.3). The immunization protection ratio of both fusion proteins in neonatal mice was 86.4% and 91.7%, respectively. These findings laid a foundation for further developing a high-efficiency subunit vaccine of BRV VP7 gene.
2.Introduction
3.Material and Methods
3.1 Cell cultures of Bovine rotavirus
3.2 Primers designs and synthesis
3.3 RNA extractions and cDNA synthesis
3.4 RT-PCR of theVP7 gene
3.5 Ligation, transformation and induction of VP7 gene and pEASY-T3 vector
3.5 Construction and PCR of recombinant plasmid
3.6 Prokaryotic expression plasmid constructions, identification and sequencing
3.7 Protein expressions and purity identification
3.8 Inoculation of mice with recombinant proteins
3.9 Protection tests of neonatal mice
4.Results
4.1 PCR amplification of BRV VP7 gene
4.2 PCR amplification of LTB gene
4.3 Identification of the recombinant pEASY-T3-VP7 plasmid and pET32a-VP7
4.4 SDS-PAGE analysis and purification
4.5 Detection of serum BRV VP7 IgG antibody
4.6 Protection efficiency of fusion proteins inoculation in neonatal mice
5.Discussion
6.Conclusion
7.Acknowledgement
8.References
Key words
VP7 gene; Bovine rotavirus; Prokaryotic Expression; Vaccine.
Introduction
Group A rotaviruses (RVA) are deemed to be important viral diarrheal agents in infants and young animals, including calves. The numbers of the rotavirus-associated mortality were estimated to be 453000 in 2008 [1, 2]. The numbers of deaths were particularly high in the developing countries [3]. Even in the developed countries, rotavirus remains an important cause of morbidity.
Rotaviruses, approximately 70nm diameter in size, are classified into at least eight different groups according to the serological reactivity and genetic variability of VP6 [4] , and consist of three capsid layers including the outer structural proteins (VP7 and VP4), middle structural protein (VP6) and inner core structural proteins (VP 1, 2 and 3). VP4 (encoded by gene segment 4) and VP7 (encoded by gene segment 7, 8 or 9 depending on the strain), both of which are independently responsible for virus neutralization [5,6]. The neutralization specificity related to VP7 is referred to as the G serotype (for glycoprotein), and that associated with VP4 is referred to as the P serotype (for protease-sensitive protein) [7]. Up to date, the rotavirus species comprises at least 27 G types (according to the nt sequence of VP7) and 37 P types (according to the nt sequence of VP4) [8, 9].
VP7 protein (glycoprotein) and VP4 protein (for protease-sensitive protein) elicit the production of neutralizing antibodies, define the antigenic specificities, referred to as G type (glycoprotein) and P type (protease-sensitive protein), respectively, and are the major antigens neutralizing immune responses during rotavirus infections [10].
The rotavirus VP7 gene is highly conservative at both ends of open reading frame (ORF), such it is feasible to detect rotavirus serotypes utilizing the rotavirus specific primers. VP7 has been shown to be involved in the early interactions with cell-surface molecules, during the rotavirus entry process [11, 12].
VP7, a calcium-binding protein, plays a major role in the stability and morphology and virus particle formation of rotavirus. Previous studies reported that recombinant VP7 protein promoted the antibody production to protect the animals from rotavirus infection [13, 14]. Although VP7 produce neutralizing antibodies, the role these antibodies play in effective immunity is still unclear.
The real time fluorescence quantitative PCR (qPCR) is not only fast and accurate, and also can test against different target sequences. Its specificity is very strong [15, 16]. Its ability to quantitatively and specifically detect genes has been invaluable for both research and diagnostic applications [17]. So far, little information regarding real time fluorescence quantitative PCR (qPCR) utilized to detect RV VP7 gene has been known.
Vaccination is an optimum measure to prevent BRV infection. But there is no such vaccine up to date. World health organization (WHO) has determined the development of a rotavirus vaccine as a priority project in the future [18]. It is difficult to develop a rotavirus vaccine because the VP7 epitope is conformation and complex structure. Nowadays very little research on BRV VP7 vaccine has been performed [19, 20].
The aim of the present study was to express VP7 Gene of BRV in prokaryotic cells, construct successfully a genetically engineered bacteria for expressing VP7 protein within BL21 (DE3) cells, and also to produce the fusion protein of pEASY-T3-VP7, furthermore to testify to the protection efficiency of fusion proteins inoculation. The findings hope to serve as a scientific foundation for the further development of genetic engineering vaccine of bovine rotavirus.
The strain GSB01 of bovine rotavirus isolated in our laboratory from the fecal samples, collected from 195 Holstein calves (1-30 days old) with diarrhea [21, 22] and the neonatal calf diarrhea virus (NCDV) strain (AV-51, purchased from Chinese veterinary drugs supervisor institute, Beijing, China) of bovine rotavirus were adapted to MA-104 cell (Ganges River monkey kidney cells, Chinese supervision of veterinary drugs, Beijing, China) cultures. The cells were cultured for 4-6 days at 37°C in the incubator containing 5% carbon dioxide (CO2). The process stopped when the cytopathogenic effects (CPE) were greater than 90%. Then the cells were frozen and thawed 2 to 3 times. The viral supernatant was collected for RNA extraction or stored -80°C until processed.
For RT-PCR, the specific primers were designed using Primer Premier 5.0 software according to the highly conserved regions of BRV VP7 based on the deposited genome sequences of BRV VP7 in GenBank (Accession number No. GQ433985.1 and EU873015.1). The restriction enzymes of Hind III and BamH I were added at the 5’ end of the forward primer (N-terminal primer) and reverse primer (C-terminal primer), respectively. Forward primer : 5’-GTAAAGCTTTGGTATTGAATATACCAC-3’ (The underlined bases were the Hind III site). Reverse primer: 5’- GATGGATCCCCTGTTGGCCATCC -3’ (The underlined bases were the BamH III site). Primers were synthesized by Takara Bio Company (Dalian, China).
To obtain virus RNA from fecal samples, genomic dsRNA was extracted from purified BRV using a Trizol method (Beijing, China). For preparing RNA from virus-infected cell culture, 0.5 to 1.0 mL of virus-infected cell supernatant was pelleted by centrifugation at 5000 rpm for 20 min to pellet the virus-cell suspension for further total RNA extraction. Total RNA was obtained according to the manufacturer's instructions and was resuspended in DEPC-treated water and stored at -80°C until use.
PCR was performed on a 25μL system which contained 15.5μL diethylpyrocarbonate (DEPC) water, 0.5μL (10mM) deoxyribonucleotide triphosphate (dNTPs), 2.5μL 10×PCR buffer, 0.5μL Taq polymerase, 0.5μL BRV forward primer, 0.5μL BRV reverse primer, 5μL cDNA. The reaction conditions were as follows: primary denaturation for 5 min at 94°C with 35 cycles of 1min at 94°C, annealing for 30 at 50°C, and 2min at 72°C, and a final elongation of 10 min at 72°C. RNA samples were quantified using a Nanodrop spectrophotometer (Shanghai, China). The absorbance ratios of 260/280nm in all samples were greater than 1.9, indicating the high purity of RNA. Five microliters of the PCR products were assayed on 1.5% agarose gel electrophoresis (Solon, USA) containing 1×Gel Red (Hayward, USA) and then analyzed with the software CS Analyzer Ver 3.0 (Tokyo, Japan).
The cDNA was synthesized from the NCDV strain and extracted viral RNA by reverse transcription reaction and utilized for PCR amplification of the VP4 gene. Expected amplicons were 890 bp sizes.
The reverse transcription-PCR (RT-PCR) assay was performed by using total RNA from fecal samples with BRV as template [22]. The qPCR was done in accordance with manufacturer’s instructions for superscript II reverse transcriptase. Following substances were added into a 500μL Ependorff tube: 5μL of 10×PCR Buffer, 1.5μL of 50mM magnesium chloride (MgCl2), 1μL of 10mM dNTP Mix FP, RP (10 μM), 0.4μL LA Taq DNA polymerase, the first chain cDNA 2μL, with coke carbonic acid ethyl ester two (diethypyrocarbonate, DEPC) water added up to 50μL.
The conditions for qPCR were as follows: initial denaturation at 95°C for 15 min followed by 40 cycles of 94°C for 15sec, 55°C for 30 sec and 72°C for 40sec, and a final extension at 72°C for 10 min.
Five microliters of the PCR products were assayed on 1.5% agarose gel electrophoresis (Solon, USA) containing 1×Gel Red (Hayward, USA) and subsequently analyzed with the software CS analyzer Ver 3.0 (Tokyo, Japan).
After the recovered fragments of PCR products were ligated into pEASY-T3 vector and cloned, they were transformed into competent DH5α cells of Escherichia coli. Escherichia coli strain DH5α was cultured in Luria-Bertani medium (LB) containing 100 μg/ mL ampicillin and grown overnight at 37°C and 180 r/min. The transformation processes were conducted in the following reaction systems, which consisted of 1μL pEASY-T3 vector, 3μL PCR products, total reaction volume of 5μL. The ligation process was completed at room temperature for 5 min.
Four microliters (4μL) reaction compounds were added slowly to 100μL competent DH5α cells of E. Coli, incubated in ice for 30min, then bathed in 42°C water for 90sec, then bathed once more on ice for 5min. 900μL Luria-Bertani media (LB media) were added. The bacteria liquid was cultured in the incubator at 37°C, and rejuvenated for 1h. Bacteria liquid was induced on the plate coated with LB/Amp (50μg/mL) and isopropyl β-D-1-Thiogalactopyranoside (IPTG). Thereafter they were cultured at 37°C for 12-20h. The recombinant pEASY-T3-VP7 was then constructed.
The B subunit gene of Escherichia coli heat-labile enterotoxin (LTB, Accession number No. : M17873.1) was amplified with the forward preimer (5′-GCCGGATCCATGGCTCCTCAGTCTATTACAGAACTATG). The PCR cycle parameters were as follows: initial denaturation for 5 min at 94°C followed by 32 cycles consisting of denaturation for 45 sec at 94°C, annealing for 45sec at 53°C, extension for 45sec at 72°C and final extension for 10 min at 72°C. The amplification products were assayed on 1.2% agarose gel electrophoresis (Amresco, Solon, USA).
The ligation and transformation of LTB (lymphotoxin beta) with pEASY-T3 vector were performed as described above.
The selected single colony of bacterium was inoculated into LB media containing 200μg/mL of ampicillin and cultured with a slow oscillation at 37°C for 12 to 14h. Then the plasmids were extracted with the pure plasmid mini Kit (Beijing, China). The recombinant pEASY-T3-VP7 plasmid was digested with both restriction endonuclease of Hind III and BamH I (Dalian, China), respectively. The products were assayed on 1.5% agarose gel electrophoresis (Solon, USA). The fragments were amplified with the forward primer and reverse primer using the extracted recombinant plasmid as template. The amplification conditions were as the same described above. The sequencing was fulfilled by Sangon Biotech (Shanghai, China).
The recombinant pEASY-T3-VP7 plasmid and pET32a vector for prokaryotic expression were digested with both restriction endonuclease of Hind III and BamH I (Dalian, China), respectively. The fragments were recovered and ligated according to the manufacturer’s instruction of T4-DNA ligase (Dalian, China), then stayed overnight at 16°C. The ligation products were transformed into the BL21 (DE3) competent cells of E. Coli. Selected colonies were identified by PCR and digested with both Hind III and BamH I. The strain of positive clones were sequenced and preserved.
The recombinant genetically engineered bacteria were cultured in Luria-Bertani media (LB media) containing 200μg/mL of ampicillin up to 0.6 of the absorbance value (OD600), then added into 1.0mmol/L IPTG and induced at 30°C for 6h. Bacteria liquid was collected.
The recombinant proteins were identified on the SDS-PAGE at constant voltage 120V. Fifty milliliters (50mL) of the transferred bacteria were induced and cultured in accordance with the method mentioned above. 20mL of bacterial supernatant was purified in the QIAexpressionistTM purification system labeled with His (Beijing, China) according to the instructions. The purified recombinant protein was subjected to further SDS-PAGE analysis.
Sixty mice of 10-weeks-old with a body weight of 46.38±2.95g, were bought from Experiment Animal Center, Lanzhou University (License No. SCXK (Gansu) 2005-0007), were randomly divided into three groups (n=20). Mice in group A were used for the control group of His-Tag prokaryotic expression vector. Mice in group B were inoculated with PEASY-T3-VP7 fusion protein. Mice in group C were inoculated with the fusion protein of VP7- TB.
When the purified protein was emulsified with adjuvant at a ratio of 1:1, the mice were intramuscularly injected with this emulsion every two weeks for three times. Blood samples were taken after a week of the last injection from the orbital cavity of each mouse. Serum IgG antibody titers were detected with Mlbio® Elisa Kit for mice (Shanghai, China), according to the manufacturer’s instruction.
All mice were raised in subgroup and kept in mice cages equipped with automatic water dispensers under the same conditions in the stable room maintained at 22-24°C and 30% to 50% relative humidity, with a controlled 12-hour light-dark cycle. Mice received a commercial diet (Lanzhou, China), depending on their physiological condition. Water was supplied ad libitum. All animal procedures were carried out in strict accordance with the Gansu province committee of experimental animal care and use of China, which meets the European Union Directive 2010/63/EU on the protection of animals used for scientific purposes.
Sixty six neonatal mice (3 to 4 days old) born from the female mice were randomly allocated into three groups according to described above. All neonatal mice were intraperitoneally injected with 100μL strain GSB01 of bovine rotavirus referring to the early reports [23, 24]. Diarrhea symptoms were observed and death numbers were calculated after 3-7 days. All neonatal mice were raised under the same conditions by using the same methods described above. All procedures referring to animal treatments were approved by Gansu province committee of experimental animal care and use of China.
Results
A band of 950bp was amplified on Agarose gel electrophoresis, which was consistent with the expected size (Figure 1).
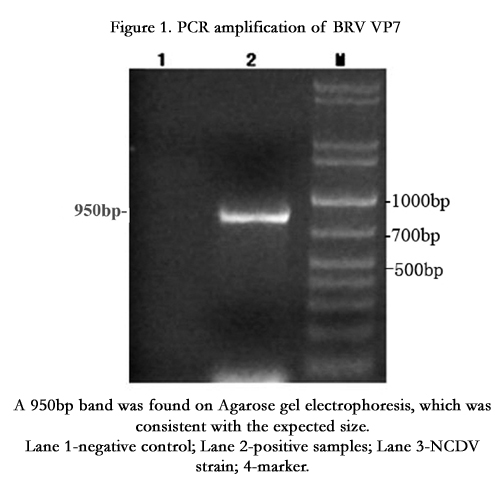
Figure 1. PCR amplification of BRV VP7
A 950bp band was found on Agarose gel electrophoresis, which was consistent with the expected size.
Lane 1-negative control; Lane 2-positive samples; Lane 3-NCDV strain; 4-marker.
A band of 310bp was amplified on Agarose gel electrophoresis, which was similar to the expected size (Figure 2).
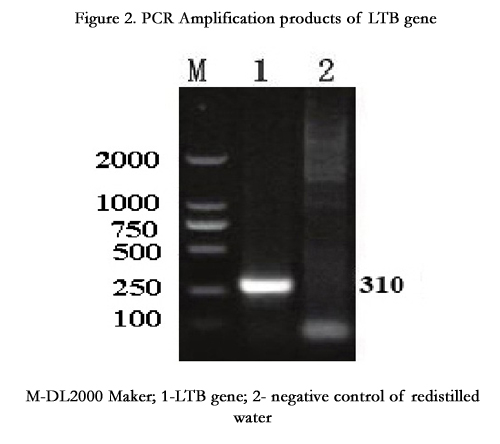
Figure 2. PCR Amplification products of LTB gene
M-DL2000 Maker; 1-LTB gene; 2- negative control of redistilled water
Following the recombinant pEASY-T3-VP7 plasmid and pET32a vector was digested with both Hind III and BamH I, respectively. The results showed that the VP7 gene had been cloned into the pEASYT3 vector. The cloned VP7 gene was introduced into pET32a vector. The recombinant pET32a-VP7 plasmid was achieved. The recombinant plasmid was named as pET32a-VP7 containing the full length VP7 cDNA.
SDS-PAGE analysis showed that the pET32a-VP7 was highly expressed in the BL21 (DE3) cells, and the molecular weights of pEASY-T3-VP7 and VP7-LTB (heat-labile enterotoxin B subunit) proteins were 42.2 kDa and 53.2 kDa, respectively. They were consistent with the expected size. (Figure 3).
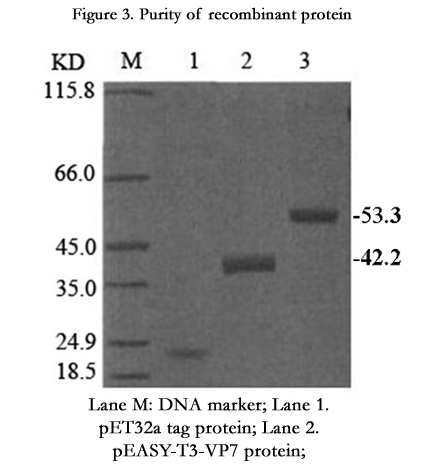
Figure 3. Purity of recombinant protein
Lane M: DNA marker; Lane 1. pET32a tag protein; Lane 2.
pEASY-T3-VP7 protein;
Lane 3. VP7-LTB protein;
The purity of the purified protein was assayed by SDS-PAGE.
The molecular weights of pEASY-T3-VP7 and VP7-LTB proteins were 42.2kD and 53.2kD, respectively.
As shown in Table 1, serum BRV VP7 IgG antibody in all mice was not detected before the fusion protein inoculation. VP7 IgG antibodies in groups A and B were significantly higher than that in group A (P<0.05 or P<0.01) following the inoculation. This illustrated that BRV VP7 gene had excellent immunogenicity. However, the VP7-LTB fusion protein could arouse a stronger immunological response.
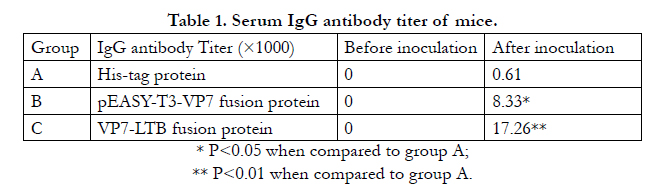

Table 1. Serum IgG antibody titer of mice.
* P<0.05 when compared to group A;
** P<0.01 when compared to group A.
Base on the clinical observation,the neonatal mice in His-tag control group showed the typical diarrhea symptoms in 3 to 7 days after intraperitoneal injection of 100μL strain GSB01 of bovine rotavirus. Mice numbers of diarrhea and death neonatal mice of pEASY-T3-VP7 and VP7-LTB fusion protein group were highly significantly less than that of His-tag control group (P<0.01). The protection efficiency of neonatal mice to pEASY-T3-VP7 and VP7-LTB fusion proteins was 86.36% and 91.67%, respectively (Table 2), which indicated that the protein was reacting with the antibody of rotavirus A.
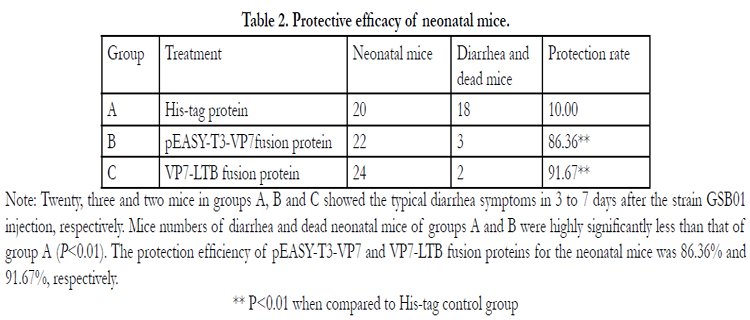
Table 2. Protective efficacy of neonatal mice.
Note: Twenty, three and two mice in groups A, B and C showed the typical diarrhea symptoms in 3 to 7 days after the strain GSB01 injection, respectively. Mice numbers of diarrhea and dead neonatal mice of groups A and B were highly significantly less than that of group A (P<0.01). The protection efficiency of pEASY-T3-VP7 and VP7-LTB fusion proteins for the neonatal mice was 86.36% and 91.67%, respectively.
** P<0.01 when compared to His-tag control group
Discussion
Rotaviruses are the major cause of acute viral gastroenteritis in infants and young children, as well as in young animals of several species, including strains of bovine, equine, porcine and canine origin [25, 26]. Worldwide prevalence rates in BRV infection range from 7% to 94%, with an average of 30%-40% [27]. BRV VP7 gene plays an important role in the stability and morphology and virus particle formation of rotavirus and takes part in the initial interactions with cell-surface molecules, during the rotavirus entry process [12]. Previous studies demonstrated that recombinant VP7 protein promoted the antibody production [13].
Vaccination is an effective measure for preventing from rotavirus infection in calves. At present, the conventional vaccines (including the live attenuated vaccine and virulent virus inactivated vaccine) are widely used [18, 28]. However, there are a lot of failures during vaccination because of very unstable of the attenuated vaccine and insufficient immune responses stimulated by the inactivated vaccine [29]. Current inactivated and attenuated vaccines have been limited owing to their high costs and inconvenience [30]. As such, it is urgently necessary to develop a new vaccine which could produce high protection efficiency [31, 32]. Successful expression of VP4 or VP7 proteins in a highly immunogenic form would be a first step towards making an effective subunit vaccine to protect against rotavirus infection [33].
The DNA vaccine is characterized with many advantages of the pure antigen, less side-effects, easy preparation, and will become the main methods of preventing BRV infection. It is a hot topic field of vaccine research and development [34]. However, there lacks in information regarding of genetic engineering vaccine research of BRV [23, 35].
In this study, the pEASY-T3-VP7 plasmid was obtained and introduced into pET32a vector. The pET32a-VP7 was also prepared and transformed to BL21 (DE3) competent cells of E. Coli. Immunogenicity of BRV VP7 and LTB were investigated preliminarily at the molecular level. BRV pEASY-T3-VP7 fusion protein and VP7-LTB fusion protein had been constructed and expressed successfully. In the neonatal mice infected with the strain GSB01 of bovine rotavirus, the protection ratio of recombinant pEASY-T3-VP7 and VP7-LTB fusion proteins inoculation was 86.4% and 91.7%, respectively. The findings demonstrated that the immunogenicity of VP7-LTB fusion protein was greater than that of pEASY-T3-VP7.
Conclusion
The genetically engineered bacteria were harvested. The pEASYT3- VP7 and VP7-LTB fusion proteins could promote the formation of IgG antibody in inoculated mice. The results lay a solid foundation for further studies on developing a highefficiency subunit vaccine and DNA vaccine of the VP7 gene for prevention and control of bovine rotavirus [30, 32].
Acknowledgement
The work received the support of the science and technology program project in Gansu province of China in 2007 (Grant No: 0708NKCA079 and the research and application project of agricultural biotechnology in Gansu province (Grant No: GNSW-2010-10).
References
- Tate JE, Steele AD, Bines JE, Zuber PL, Parashar UD (2012) Research priorities regarding rotavirus vaccine and intussusception: a meeting summary Vaccine 30(1): A179-A184.
- Tate JE, Haynes A, Payne DC, Cortese MM, Lopman BA, et al. (2013) Trends in national rotavirus activity before and after introduction of rotavirus vaccine into the national immunization program in the United States 2000 to 2012. Pediatr Infect Dis J 32(7): 741-744.
- Abe M, Ito N, Morikaw S, Takasu M, Murase T, et al. (2009) Molecular epidemiology of rotaviruses among healthy calves in of a novel bovine rotavirus bearing new P and G genotypes .Virus Res 144(1-2): 250-257.
- Desselberger U (2014) Rotaviruses. Virus Res.190: 75-96.
- Estes MK (2001) Rotaviruses and their replication In Fields virology (DM Knipe & Howley PM eds) Lippincott-Roven Publishers Philadelphia. 1747-1785.
- Suresh T, Rai RB, Dhama K, Sawant PM, Kumar D, et al. (2012) Determination of G and P type diversity of group A rotaviruses and detection of a new genotype from diarrhoeic calves in northern and southern states of India .Vet Pract 13(1): 1-8.
- Anthony HC, Choi MB, Monica MM (1999) Antibody-Independent Protection against Rotavirus Infection of Mice Stimulated by Intranasal Immunization with Chimeric VP4 or VP6 Protein. J Virol 73(9): 7574-7581.
- Matthijnssens J, Ciarlet M, McDonald SM, Attoui H, Bányai K, et al. (2011) Uniformity of rotavirus strain nomenclature proposed by the Rotavirus Classification Working Group (RCWG). Arch Virol. 156(8): 1397-1413.
- Rotavirus Classification Working Group (2013) 6th meeting, Valencia.
- Aminu M, Page NA, Ahmad AA Umoh JU, Dewar J, Steele AD (2010) Diversity of Rotavirus VP7 and VP4 Genotypes in Northwestern Nigeria J Infect Dis 202(1): S198-S204.
- Lopez S, Arias CF (2006) Early steps in rotavirus cell entry. Curr Top Microbiol Immunol 309: 39-66.
- Martha NC, Fanny G, Orlando A, Carlos AG (2012) Rotavirus VP4 and VP7-Derived Synthetic Peptides as Potential Substrates of Protein Disulfide Isomerase Lead to Inhibition of Rotavirus Infection. Int J Pept Res Ther 18: 373-382.
- Johnson MA, Misra RM, Lardelli M (1989) Synthesis in Escherichia coli of the major glycoprotein of human rotavirus:analysis of the antigenic regions. Gene 84(1): 73-81.
- Wang L, Huang JA, Nagesha HS (1999) Bacterial expression of the major antigenic regions of porcine rotavirus VP7 induces a neutralizing immune response in mice .Vaccine 17(20-21): 2636-2645.
- Okada N, Matsumoto Y (2002) Bovine rotavirus G and P types and sequence analysis of the VP7 gene of two G8 bovine rotaviruses from Japan.Vet Microbiol 84(4): 297-305.
- Santos N, Hoshino Y (2005) Global distribution of rotavirus serotypes/ genotypes and its implication for the development and implementation of an effective rotavirus vaccine Rev Med Virol 15(1): 29-56.
- Schweitzer B, Kingsmore S (2001) Combining nucleic acid amplification and detection. Curr Opin Biotechnol 12(1): 21-27.
- Roger IG, Umesh DP, Joseph SB, Reina T, Thea KF, et al. (2006) Rotavirus vaccines: current prospects and future challenges. Lancet 368: 323-332.
- Gouvea V, Glass RI, Woods P, Taniguchi K, Clarke HF, et al. (1990) Polymerase chain reaction amplification and typing of rotavirus nucleic acid from stool specimens. J Clin Microbiol 28(2): 276-282.
- Mohan S, Manoharan P, Pachaikani R (2006) Genotyping of rotavirus of neonatal calves by nested-multiplex PCR in India .Veterinarski Arhiv 76(6): 497-505.
- Wei Suocheng, Feng Ruofei, Gong Zhuandi, Tian Fengling (2010) Researches of Isolation and Cell Cultivation of Bovine Rotavirus in Vero and MA-104 Cells. Journal of Northwest University for Nationalities 31 (4): 71-75.
- Wei S, Gong Z, Che T, Ayimu G, Tian F (2013) Genotyping of calves rotavirus in China by reverse transcription polymerase chain reaction. J Virol Methods 189(1): 36-40.
- Wang Zhiming, Meng Yajuan, He Cheng, Gao Jian (2013) Status and Progress of Research on Mucosal Vaccines. Chin Pharm J 49(1): 1-6.
- Hu J, Wang C (2008) Expression and Immunogenicity Analysis of Recombinant Plasmid pW425et-Vp7 of Porcine Rotavirus A in Lactobacillus. Acta Microbiologica Sinica 48(11): 1514-1519.
- Yang S, He H, Yang H, Wang C, GaoY, et al. (2011) Genetic engineering subunit vaccine of group A bovine rotavirus. Chinese J Zoonoses 27: 505-510.
- Wei suocheng (2005) Epidemiology and integrated control of animal rotavirus disease Animal Husbandry and Veterinary in Gansu 27(3): 27-28
- Fukai K, Takahashi T,Tajima K, Koike S, Iwane K, et al. (2007) Molecular characterization of a novel bovine group A rotavirus. Vet Microbiol 20: 217-224
- van Der Heide R, Koopmans MP, Shekary N, Houwers DJ, van Duynhoven YT, van der Poel WH (2005) Molecular characterizations of human and animal group a rotaviruses in the Netherlands. J Clin Microbiol 43: 669-675.
- Lokeshwar MR, Bhave S, Gupta A, Goyal VK, Walia A (2013) Immunogenicity and safety of the pentavalent human-bovine (WC3) reassortant rotavirus vaccine (PRV) in Indian infants. Hum Vaccin Immunother 9 (1):172-176.
- Clark HF, Offit PA, Plotkin SA, Heaton PM (2006) The New Pentavalent Rotavirus Vaccine Composed of Bovine (Strain WC3) -Human Rotavirus Reassortants. Pediatric Infect Dis J 25 (7): 577-583.
- Chen Qian, Zhang Ying, Qian Zhi-yong (2014) Case-control Study on Effect of Oral Rotavirus Attenuated Live Vaccine against Rotavirus Diarrhea Chinese Journal of Vaccines and Immunization 20 (1): 55-57
- Wei Z, Jianbao D, Takeshi H, Yoshitaka G, Masuo S (2011) Rapid and sensitive detection of bovine coronavirus and group A bovine rotavirus from fecal samples by using one-step duplex RT-PCR assay. J Vet Med Sci 73: 531-534.
- Gökçe G, Ran Z, Lennart H, Harold M (2014) Engineered Lactobacillus rhamnosus GG expressing IgG-binding domains of protein G: Capture of hyperimmune bovine colostrum antibodies and protection against diarrhea in a mouse pup rotavirus infection model. Vaccine 32 (4): 470-477.
- Nazlina I, Malcolm A M (2001) High-level expression of bovine rotavirus VP4 protein in Escherichia coli using a hepatitis B core antigen vector Biotechnology Letters 23: 643-646
- Rajtak U, Leonard N, Bolton D, Fanning S (2011) A real-time multiplex SYBR Green I polymerase chain reaction assay for rapid screening of salmonella serotypes prevalent in the European Union. Foodborne Pathog Dis 8 (7): 769-780.
- Jiang Baoming, Yang Xiaoming, Xu Deqi (2012) The current status and development trend of rotavirus vaccine. China Journal of Biologicals 25 (2):251-253.





