Combined Duodenal Atresia and Pure Esophageal Atresia
Alaish SM
Assistant Professor, Department of Surgery, University of Maryland, School of Medicine, USA.
*Corresponding Author
Samuel M. Alaish,
Assistant Professor,
Department of Surgery,
University of Maryland,
School of Medicine, USA.
Tel: (410) 328-5730
Fax: (410) 328-0652
E-mail: salaish@smail.umaryland.edu
Article Type: Case Report
Received: October 29, 2014; Accepted: February 22, 2015; Published: February 23, 2015
Citation: Alaish SM (2015) Combined Duodenal Atresia and Pure Esophageal Atresia. Int J Surg Res 2(1) 5-7. doi:dx.doi.org/10.19070/2379-156X-150002
Copyright: Alaish SM© 2014. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
Abstract
Combined duodenal atresia and pure esophageal atresia is a rare combination which has been reported only 13 times previously in the literature. Although this pair of congenital anomalies is amenable to current treatment strategies, it is often associated with additional defects which lead to the baby’s demise. We present a newborn prenatally diagnosed with trisomy 21, duodenal atresia and an AV canal defect. Postnatally, these diagnoses were confirmed, and the baby was also found to have pure esophageal atresia. Following a successful, staged repair of his duodenal atresia and pure esophageal atresia, the baby developed severe tracheobronchomalacia. At that point, the parents decided to withdraw care rather than embark on airway stent placement followed by cardiac surgery. This report highlights two of the most challenging aspects of pediatric surgery: 1) severe newborn anomalies requiring sound planning strategies and technical expertise and 2) an intimate relationship with the parents, so that the care can be individualized for the wishes of the family.
2.Introduction
3.Case Presentation
4.Discussion
5.References
Keywords
Duodenal Atresia; Pure Esophageal Atresia; Tracheobronchomalacia; Pediatric Surgery; Withdraw Intensive Care.
Introduction
Duodenal atresia is not an uncommon congenital anomaly, occurring in 1 in 6,000 to 1 in 10,000 live births [1]; it is also frequently associated with trisomy 21, which carries a high rate of congenital heart defects. On the other hand, pure esophageal atresia alone is rare, occurring in 1 in 36,000 births [2]. The present case is now the 14th reported case of combined duodenal atresia and pure esophageal atresia in the literature. Despite successful surgical treatment strategies for these two conditions, overall survival has been a dismal 50% [3] due to other associated anomalies.
Case Presentation
A 2.68 kg white male infant with a prenatal diagnosis of duodenal atresia, AV septal defect and possible Down syndrome was born via a Cesarean section at 36 weeks’ gestational age to a 27-yearold gravida 2, para 1 mother whose pregnancy was complicated by polyhydramnios. Apgars were 9 at 1 minute and 9 at 5 minutes. The baby was pink and vigorous upon arrival in the NICU and was stable on room air. Of note, the baby did not pass meconium, and a nasogastric tube could not be passed. Physical examination demonstrated a facies consistent with Down syndrome [later confirmed by karyotype], an abdominal examination demonstrating a large, ballotable mass extending from one side of the mid-abdomen to the other, and a rectal exam met with only white mucus in the vault. The initial postnatal chest and abdominal radiograph demonstrated a nasogastric tube coiled in the upper esophagus and a gasless abdomen, thus consistent with a diagnosis of pure esophageal atresia (Figure. 1). Postnatal echocardiography confirmed the prenatal diagnosis of an AV canal defect and also showed a patent ductus arteriosus. An abdominal ultrasound yielded a “double bubble” sign diagnostic of duodenal atresia (Figure. 2).
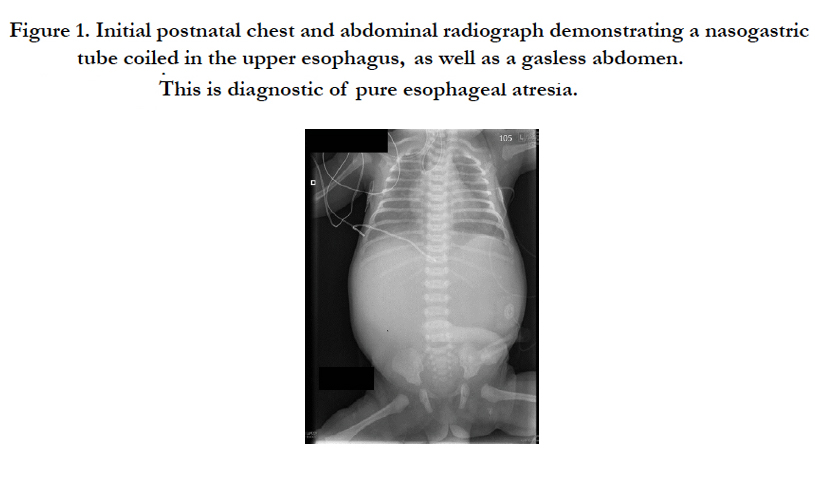
Figure 1. Initial postnatal chest and abdominal radiograph demonstrating a nasogastric tube coiled in the upper esophagus, as well as a gasless abdomen. This is diagnostic of pure esophageal atresia.
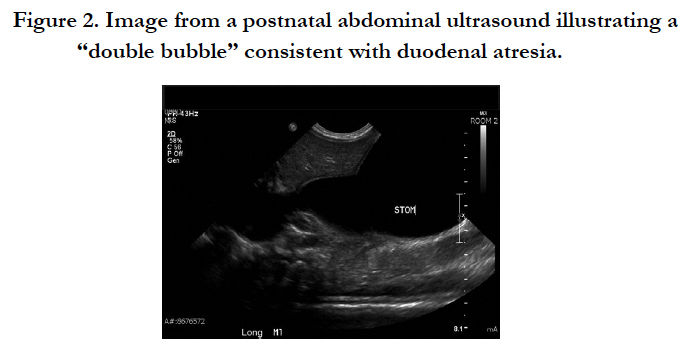
Figure 2. Image from a postnatal abdominal ultrasound illustrating a “double bubble” consistent with duodenal atresia.
Following 24 hours of acclimation, the baby was taken to the operating room and underwent an exploratory laparotomy through a right-sided transverse upper abdominal incision. After a Kocher maneuver, the massive stomach and proximal duodenum were well-visualized. The baby had normal intestinal rotation. The stomach was decompressed using a 20-gauge needle, and 240 ccs of nonbilious fluid was aspirated. The distal duodenum could now be seen, and a type I duodenal atresia was found. In preparation for a web resection, a longitudinal incision was made in the dilated proximal duodenum. Through this opening, a 20-Fr bougie was placed into the stomach and advanced into the distal esophageal pouch. Under fluoroscopy, we were able to measure the gap between the proximal esophageal pouch, which had a nasogastric tube in place, and the distal esophageal pouch with the bougie. The gap measured 3 vertebral bodies in length (Figure. 3). The bougie was removed. Close inspection of the duodenal web revealed it to be intimately associated with the ampulla of Vater, so rather than a web resection, we incised the distal duodenum and performed a side-to-side duodenoduodenostomy. Prior to the anastomosis, the distal duodenum was interrogated with a red rubber catheter and saline; no other obstructions were found. A Stamm gastrostomy was done prior to closing. The baby recovered from this operation uneventfully and tolerated full enteral feeds via his gastrostomy.
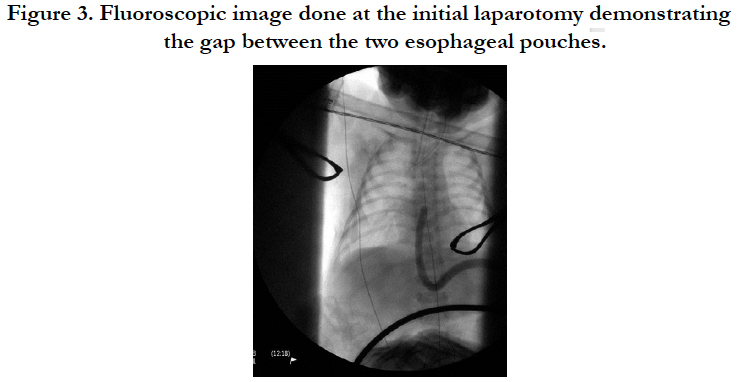
Figure 3. Fluoroscopic image done at the initial laparotomy demonstrating the gap between the two esophageal pouches.
At one month of age, the baby was taken to the fluoroscopy suite, where the gap between the two esophageal pouches was again measured. With a nasogastric tube in the upper pouch and a Hegar dilator passed through the gastrostomy, the gap measured less than 2 vertebral bodies (Figure. 4). On day of life 36, the baby returned to the operating room and underwent a rigid bronchoscopy which ruled out a proximal tracheoesophageal fistula. We then proceeded with a right posterolateral thoracotomy and fashioned an esophagoesophagostomy (Figure. 5). Following this operation, the baby failed multiple attempts at extubation and required a tracheostomy. Rigid bronchoscopy diagnosed severe tracheobronchomalacia (Video 1). After extensive discussions with the family, the family opted to withdraw care rather than continue with airway stent placement and future cardiac surgery. Simply, they felt their child had been through enough.
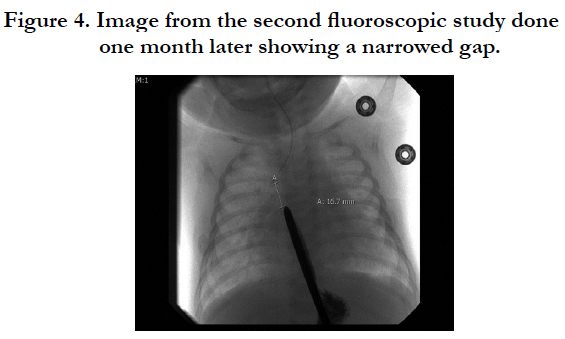
Figure 4. Image from the second fluoroscopic study done one month later showing a narrowed gap.
Figure 5. Intraoperative photograph at the time of the thoracotomy showing the completed esophagoesophagostomy. The head of the patient is to the left.
Discussion
Our patient is the 14th reported case of combined duodenal atresia and pure esophageal atresia. Two of these were aborted fetuses. Of the 12 remaining live births, there have been only 7 survivors. Additional associated anomalies previously reported include extra ribs, choledochal cyst, biliary and pancreatic ductal atresia, and unilateral lung agenesis. Remarkedly, the infant with unilateral lung agenesis in addition to combined duodenal atresia and pure esophageal atresia is a recent survivor [4]. Also of note, Down syndrome was diagnosed in 4 of the 12 babies, and only 1 survived. Down syndrome is highly associated with congenital heart disease, and this likely impacted survival, as it did in our case. In a review of 670 patients [5] with esophageal atresia, chromosomal abnormalities were recognized in 35 (5.2%). Trisomy 18 was the most common abnormality with trisomy 21 second. Of the 12 patients with Down syndrome, 6 had pure esophageal atresia (50%) compared to a 6% incidence of pure esophageal atresia in the rest of the patients. As expected, Down syndrome patients were also more likely to have congenital heart disease, duodenal atresia and Hirschsprung disease. Overall mortality in this series was 7% for patients without a chromosomal abnormality and 70% for patients with a chromosomal abnormality.
The decision by the parents to withdraw intensive care for their baby was set in motion following failed extubation attempts due to the severe tracheobronchomalacia. Unfortunately, the use of a long Shiley tracheostomy tube could only stent open the trachea but not the right mainstem bronchus without occluding the left mainstem bronchus. Similarly, a standard aortopexy would not have kept the right mainstem bronchus open. The use of airway stents to manage tracheobronchomalacia in both premature and young term infants has been reported [6, 7] and was discussed with the family; however, the use of airway stents is not without complications including restenosis by granulation tissue, infection, failure of tracheal growth, migration, tracheal erosion or penetration to the great vessels [8]. Furthermore, the midrange and long-term outcomes from airway stent management in this very young infant population is not well-described. The uncertainty surrounding this modality coupled with the infant’s looming cardiac surgery led the parents to decide that their child should just receive comfort care. The parents of our patient came to their decision after placing the highest priorities on quality of life, likelihood of improvement and perception of their child’s pain. These are the exact same priorities previously noted in a study of parental perspectives on end-of-life care in the pediatric intensive care unit [9].
In summary, this case highlights the essence of pediatric surgery. A pediatric surgeon must possess both sound clinical judgment to plan care and fine technical skills to correct complex neonatal anomalies. At the same time, the surgeon must balance the patient’s treatment strategy with the needs of the family through active dialogue, so that the care is individualized and meets the wishes of the family.
References
- Fonkalsrud EW, DeLorimier AA, Hays DM (1969) Congenital atresia and stenosis of the duodenum: a review compiled from the members of the Surgical Section of the American Academy of Pediatrics. Pediatrics 43(1):79-83.
- Spitz L (2007) Oesophageal atresia. Orphanet Journal of Rare Diseases 2(24):1-13.
- Pameijer CR, Hubbard AM, Coleman B, Flake AW (2000) Combined pure esophageal atresia, duodenal atresia, biliary atresia, and pancreatic ductal atresia: prenatal diagnostic features and review of the literature. J PediatrSurg 35(5):745-747.
- Downard CD, Kim HB, Laningham F, Fishman SJ (2004) Esophageal atresia, duodenal atresia and unilateral lung agenesis: a case report. J PediatrSurg 39(8):1283-1285.
- Beasley S, Allen M, Myers N (1997) The effects of Down syndrome and other chromosomal abnormalities on survival and management in oesophageal atresia. PediatrSurgInt 12(8): 550-551.
- Torer B, Gulcan H, Oguzkurt L, Oguzkurt P, Tarcan A (2008) Use of balloon-expandable metallic stent in a premature infant with congenital tracheobronchial stenosis. PediatrPulmonol 43(4):414-417.
- Sauvat F, Michel JL, Harper L, Mirabile L, Hoi RW, et al.(2011) Successful management of congenital bronchial stenosis using an expandable stent. J PediatrSurg 47(1): E1-E4.
- Anton-Pacheco JL, Cabezali D, Tejedor R, Lopez M, Luna C, et al.(2008) Eur J CardiothorSurg 33: 1069-1075.
- Meyer EC, Burns JP, Griffith JL, and Truog RD (2002) Parental perspectives on end-of-life care in the pediatric intensive care unit. Crit Care Med 30(1):226-231.





