A Type 3 Klippel Feil Syndrome Case With Atrial Septal Defect and Atrial Septal Aneurysm
Karaağaç AT*, Yildirim AI
Kartal KoŞuyolu Research and Training Hospital, Pediatry, Istanbul, Turkey.
*Corresponding Author
Aysu Türkmen Karaağaç,
Kartal Koşuyolu Research and Training Hospital,
Denizer Cad, Cevizli kavşağı, No:2, 34846,
Kartal, Istanbul, Turkey.
Tel: 0(216)5001500-1101
E-mail: aysukaraagac@gmail.com
Article Type : Case Study
Received: January 20, 2016; Accepted: January 20, 2016; Published: January 22, 2016
Citation: Karaagac AT, Yildirim AI (2016) A Type 3 Klippel Feil Syndrome Case With Atrial Septal Defect and Atrial Septal Aneurysm. Int J Pediat Health Care Adv. 3(1), 13-16.
Copyright: Karaagac AT© 2016. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
Abstract
Klippel Feil syndrome (KFS) is a rare congenital disorder characterized by short neck due to abnormal fusion of cervical vertebrae, limited head and neck motion and low posterior hairline. We present a rare case of type 3 KFS with the fusion of cervical and thoracic vertebrae, cervical hemivertebrae anomaly, Sprengel’s deformity, rib fusion anomaly, accompanied by atrial septal defect/septal aneurysm, thereby reviewing the literature about this syndrome.
2.Introduction
3.Case Presentation
4.Discussion
5.Conclusion
6.References
Keywords
Cervical Vertebrae; Fusion Anomaly; Cardiac Defect; Klippel-Feil Syndrome.
Introduction
KFS, first described by Maurice Klippel and Andre Feil in 1912, is believed to result from faulty segmentation of the long axis of developing embryo dring the second to eighth weeks of gestation [1]. Besides its classical triad, a variety of clinical symptoms such as scoliosis, rib anomalies, raised scapula (Sprengel’s deformity), facial asymmetry, cardiac, auditory or urinary anomalies may accompany KFS [1, 2]. Its incidence was reported as 0.2-0.7 cases per 1000 people [3].
Case Presentation
Ş.Y, 7-year-old female was referred to our hospital for cardiac surgery. She was born with a ceserian section, 2700 gr in weight and 46 cm. in height. She remained in the neonatal intensive care unit for 10 days due to the respiratory difficulty. Her mother and father were both 28 years old and 3rd degree relatives. They had no physical abnormality or history of any chronical illness. The gestational follow up of the mother was normal. She had no history of cigarette smoking, drug or alcohol use.
On the physical examination in our inpatient clinic, Ş.Y had facial asymmetry, prominent forehead and occiput, low posterior hairline, short tilted neck, limited neck motion, scoliosis, kyphosis and a raised scapula. (Figure 1) Her weight was 15 kg(<3rd percentile) and height was 105 cm.(3rd percentile). There was a concavity on the left thoracic region of her chest (Figure 2). Her respiratory sounds were normal and cardiac auscultation revealed 2/6 systolic murmur on the pulmonic area with 86 heart beats per minute. Her oxygen saturation was 96% in the room air. The rest of the systemic examination was normal. On her laboratory test results there was no abnormality except for a moderate degree of anemia with a hemoglobin level of 9 gr/dl and hematocrit (Htc) %26. Her 3 dimensional cervical computed tomography (CT) showed multiple hemivertebrae anomalies, fusion of cervical vertebrae and disruption in the atlantoaxial junction (Figure 3). Spinal magnetic resonance imaging (MRI) showed multiple fusion anomalies in the cervical and upper thoracic vertebrae (Figure 4). The plain radiography of the chest revealed rib fusion anomaly on the left thoracic region (Figure 5). Echocardiographic examination demonstrated atrial septal defect, atrial septal aneurysm, tricuspid regurgitation and dilatation in the right heart chambers. Abdominal ultrasonography was normal except for a mild hepatomegaly. Fluorescence in situ hybridization (FISH) analysis performed by D22S75(Aquarius/Cytocell) probe showed no chromosome abnormality in our case and her parents and the karyotype analysis was normal (46,XX).

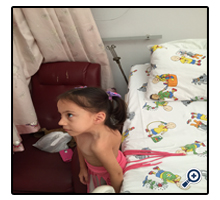
Figure 1. The prominent forehead, low posterior hairline, short tilted neck, scoliosis and a raised scapula in our KFS case.
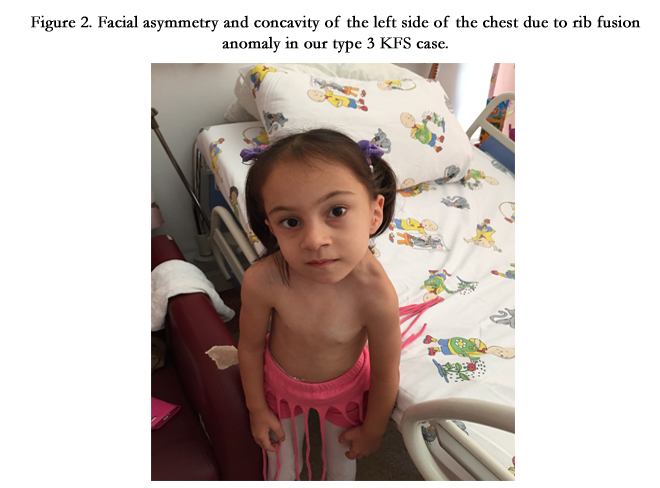
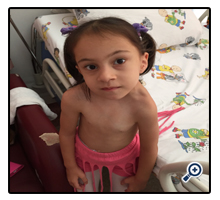
Figure 2. Facial asymmetry and concavity of the left side of the chest due to rib fusion anomaly in our type 3 KFS case.
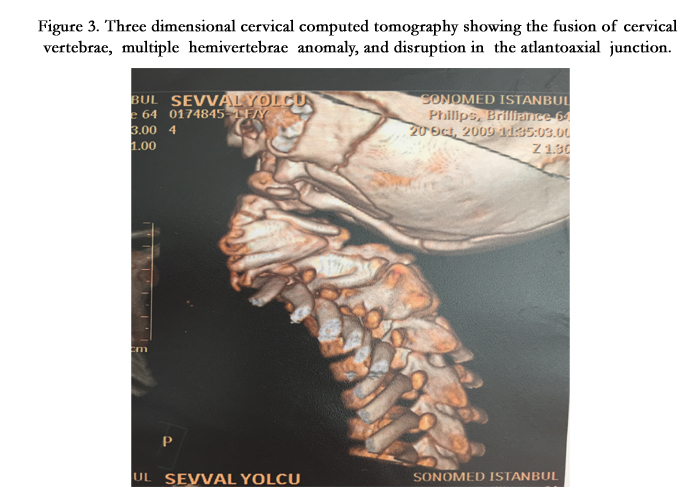

Figure 3. Three dimensional cervical computed tomography showing the fusion of cervical vertebrae, multiple hemivertebrae anomaly, and disruption in the atlantoaxial junction.
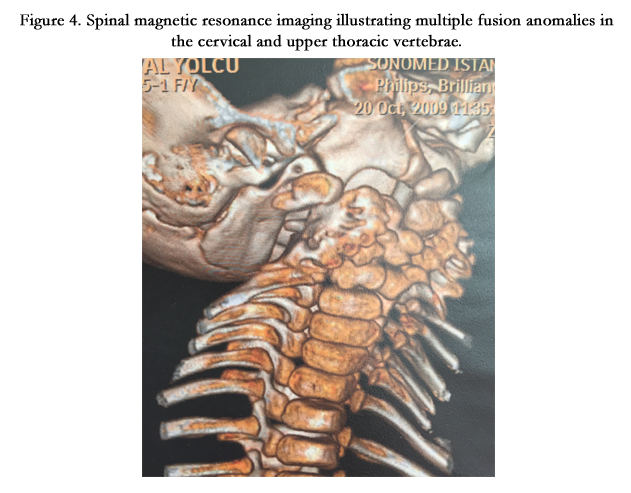

Figure 4. Spinal magnetic resonance imaging illustrating multiple fusion anomalies in the cervical and upper thoracic vertebrae.
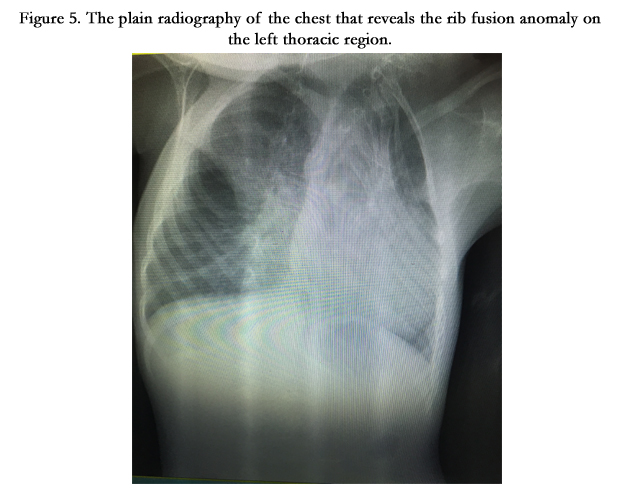
Figure 5. The plain radiography of the chest that reveals the rib fusion anomaly on the left thoracic region.
Discussion
Klippel Feil is a rare syndrome that can be diagnosed incidentally at any age throughout life due to a wide range in severity of its symptoms. Individuals with upper cervical spine anomalies usually present at earlier ages than those with lower cervical spine involvement. 40-50% of the cases present with short neck, decreased range of motion of the cervical spine and low posterior hairline [2]. Rotation of the neck rather than the flexion or extension is limited. Studies have showed that 20-40% of the affected individuals have torticollis, 40-50% scoliosis and 30% additional skeletal deformities as Sprengel’s deformity (elevated scapula), syringomyelia and/or fusion of ribs [3, 4]. Our patient had a tilted neck, torticollis, cervical rotation anomaly, Sprengel’s deformity, rib fusion anomaly, severe scoliosis and a moderate degree kyphosis.
Feil classified the syndrome into 3 categories: Type 1 includes the cases with fusion of cervical vertebrae, type 2 cases have fusion of cervical and thoracic vertebrae with associated cervical hemivertebrae (incomplete development of one half of any vertebra) anomaly and abnormal fusion of atlantoaxial joint with the occiput. Type 3 KFS is characterized by the fusion of cervical, thoracic and/or lumbar vertebrae with associated rib anomalies [5-7]. As her cervical 3D CT and spinal MRI showed fusion of the occiput, upper cervical and upper thoacic vertebrae with cervical hemivertebrae anomaly and plain radiography of the chest revealed fusion of the ribs on the left side of the thorax, our case was classified as type 3 KFS. Gray et al. found that fusion of the occiput with the first, second and third vertebrae produces most of the neurologic symptoms especially in the first decade of life. Therefore, these individuals should avoid from activities that could lead to cervical trauma, which may result in hemi or quadriplegia [8, 9]. As our case was 7 year-old type 3 KFS case with a great risk of neurologic impairment, neurosurgeons have planned close follow up and operation to correct the compression and associated vertebral instability if necessary. 25-35 % of KFS cases may suffer from facial asymmetry, audotory problems such as hearing loss or visual abnormalities as strabismus, coloboma or nystagmus [2, 4]. Our case had minimal facial asymmetry, but no visual or audotory problems.
Congenital heart defects may sometimes (4%-14%) accompany KFS. The most common cardiac anomalies associated with KFS are ventricular septal defect and aortic arc anomalies. Zaki et al. reported a KFS case with isolated hypokinesia of the left ventricule. Elumalai et al. reported another KFS case with complete heart block that required pacemeker implantation. Therefore, cardiologic evaluation of KFS cases is essential [10 -12]. Electrocardiography was normal, but the echocardiographic examination of our case demonstrated atrial septal defect, atrial septal aneurysm and dilatation of the right side of the heart. Her cardiac defect was successfully corrected by surgery.
As the cervical vertebrae and genitourinary system differentiate at the same embryological period, urinary system abnormalities may be encountered in 30-35% of the KFS cases, the most common being unilateral renal agenesis [13]. Therefore, urinary ultrasonography and intravenous pyelography, if necessary, should be performed to the KFS patients to eliminate renal anomalies including renal agenesis, double collecting system, horseshoe shaped kidneys, renal ectopia or hydronephrosis. Renal anomaly was not detected in the abdominal ultrasonography of our case. The etiology of KFS and its associations haven’t been clarified yet. Most of the KFS cases are sporadic. In several genetic studies it has been reported that the mutations of MEOX1 gene that codes for mesenchyme may lead to a recessive subtype of KFS. However, Gray et al. found little correlation with inheritance [14, 15]. The FISH analysis of our case and her parents didn’t reveal chromosomal abnormality, however we think that genetic counseling should be provided to the suspected KFS cases and their parents for subsequent pregnancies.
Conclusion
Klippel-Feil is a rare syndrome including multiple systemic associations with a wide range of severity. Therefore, it requires a multidisciplinary approach to diagnose, monitorize and treat its complications. In such syndromic cases it should be remembered that the earlier the diagnosis, the less the systemic impairments. Our case was a 7 year-old type 3 KFS case with highly severe vertebrae anomalies and cardiac involvement. Her cardiac anomaly was corrected, but she has being followed by neurosurgeons and orthopedicians for the surgical correction and stabilization of her vertebrae anomalies to prevent the neurologic impairment and to make her life more comfortable.
References
- Mahiroğullari M, Ozkan H, Yildirim N, Cilli F, Güdemez E (2006) Klippel- Feil syndrome and associated congenital abnormalities: Evaluation of 23 cases. Acta Orthop Traumatol Turc 40(3): 234-239.
- Naikmasur VG, Sattur AP, Kirty RN, Thakur AR (2011) Type III Klippel- Feil syndrome: case report and review of associated craniofacial anomalies.Odontology 99(2): 197-202.
- Sullivian JA (2009) Klippel-Feil syndrome. Emedicine Journal.
- Tracy MR, Dormans JP, Kusumi K (2004) Klippel-Feil syndrome: clinical features and current understanding of etiology. Clin Orthop Relat Res (424): 183-190.
- Fedala S, Mahdi HA, Aicha T, Leyla AA, Djamila M (2015) Klippel Feil Syndrome: A case report and review of literature. Int J Clin Causes Investig 6(2): 62-67.
- Samartzis DD, Herman J, Lubicky JP, Shen FH (2006) Classification of congenitally fused cervical patterns in Klippel-Feil patients: epidemiology and role in the development of cervical spine-related symptoms. Spine 31(21): E798-804.
- Erol FS, Ucler N, Yakar H (2011) The association of Chiari type III malformation and Klippel-Feil syndrome with mirror movement: a case report. Turk Neurosurg 21(4): 655-658.
- Samartzis D, Lubicky JP, Herman J, Kalluri P, Shen FH (2006) Symptomatic cervical disc herniation in a pediatric Klippel-Feil patient: the risk of neural injury associated with extensive congenitally fused vertebrae and a hypermobile segment. Spine 31(11): E335-338.
- Auerbach JD, Hosalkar HS, Kusuma SK, Wills BP, Dormans JP, et al. (2008) Spinal cord dimensions in children with Klippel-Feil syndrome: a controlled, blinded radiographic analysis with implications for neurologic outcomes. Spine 33(12): 1366-1371.
- Zaki AS, Shenoy P, Shanbag P (2010) Klippel Feil syndrome with isolated hypokinesia of the left ventricle: A rare association. Ann Pediatr Cardiol 3(1): 92-93.
- Driazhenkova IV (2005) The diastolic dysfunction of the left ventricle in patients with systemic lupus erythematosus and system scleroderma. Klin Med (Mosk) 83(7): 45-47.
- Elumalai RS, Nainar MS, Vaidyanathan K, Somasundaram G, Balasubramaniam G (2013) Congenital complete heart block in Klippel-Feil syndrome. Asian Cardiovasc Thorac Ann 21(2): 199-201.
- Moore WB, Matthews TJ, Rabinowitz R (1975) Genitourinary anomalies associated with Klippel-Feil syndrome. J Bone Joint Surg Am 57(3): 355- 357.
- Mohamed JY, Faqeih E, Alsiddiky A, Alshammari MJ, Ibrahim NA, et al. (2013) Mutations in MEOX1, encoding mesenchyme homeobox 1, cause Klippel-Feil anomaly. Am J Hum Genet 92(1): 157-161.
- Bayrakli F, Guclu B, Yakicier C, Balaban H, Kartal U, et al. (2013) Mutation in MEOX1 gene causes a recessive Klippel-Feil syndrome subtype. BMC Genetics 14: 95-96.





