A Patient with a Rare Chromosome Deletion Presents with Evidence of Spontaneous Resolution of Primary Congenital Glaucoma
Walker K
Assistant Professor, Western University of Health Sciences College of Optometry, USA.
*Corresponding Author
Kimberly Walker, OD FCOVD,
Assistant Professor,
Western University of Health Sciences College of Optometry,
USA.
Tel: 909-469-8478
Fax: 909-706-3905
E-mail: kwalker@westernu.edu
Article Type: Case Report
Received: September 25, 2014; Accepted: January 16, 2015; Published: February 04, 2015
Citation: Walker K (2015) A Patient with a Rare Chromosome Deletion Presents with Evidence of Spontaneous Resolution of Primary Congenital Glaucoma. Int J Ophthalmol Eye Res. S1:001, 1-4. doi: dx.doi.org/10.19070/2332-290X-SI01001
Copyright: Walker K© 2015. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
Abstract
Background: Deletions of chromosome 6q25 are very rare. Patients with these deletions present with variable characteristics depending on the location, size and break points of the deletion. In the literature it has been associated with developmental delays, brain abnormalities, hearing impairments, as well as other variable anomalies. There have been ocular abnormalities reported including retinal pigment irregularities and strabismus. Depending on their impairments, these patients can have a normal life expectancy. To our knowledge there have been no reports of congenital glaucoma with thischromosome deletion.
Case Report: A 19 month old Hispanic male was referred to our clinic by his infant development teacher, who noticed that he was not visually engaged during therapy. Hewas born with a rare chromosome deletion of 6q25. He is severely developmentally delayed in all areas. He also has absence seizures for which he is prescribed Keppra and Phenobarbital. He had a previous eye examination at 3 weeks of age. In reviewing those records, he presented with a chief complaint of watery eyes OU. He was diagnosed with having blond fundus with all findings being within normal limits. The plan was to follow up in 6 weeks. The parents elected not to return for their 6 week appointment, due to dissatisfaction with the previous visit. His examination at our clinic reveled large cornea’sOU, restricted visual field OS worse than OD, obvious preference for use of his right eye, HaabStriae OS>OD, andcup to disc ratios of 0.80/0.80 OD, 0.95/0.95 OS. IOP measured by Tonopen was 18 mmHg OD and 13 mmHg OS. The patient was referred for evaluation and treatment with a pediatric ophthalmologist.
Conclusion: To the best of our knowledge this is the first case that has been reported of congenital glaucoma in the presence of this rare deletion. The initial symptoms of epiphora, which can be a sign of congenital glaucoma, were not addressed in this patient’s first eye examination. While this case was an atypical presentation of congenital glaucoma, with normal IOP, there are indications that the IOP was previously elevated, indicating possible resolution of condition.
2.Background
2.1.Deletions of Chromosome 6q25
2.2.Congenital Glaucoma
3.Case Report
4.Discussion
5.Conclusion
6.References
Keywords
Congenital Glaucoma; Chromosomal Deletion.
Background
Interstitial deletions of chromosome 6q are very rare.Since the first case was reported in 1975, just over 65 cases have been reported in the literature. [1,2] Patients with 6q25 deletions present with variable characteristics depending on the location, size and break points of the deletion. Published cases of this deletion report a wide array of dysfunctions.1-6Intellectual disabilities, global developmental delays, postnatal growth retardation, feeding difficulties, gastrointestinal abnormalities, hearing impairments, dysmorphic features of the face, head (microcephaly) and genitalia, heart defects, seizures and structural brain abnormalities including dysgenesis of the corpus callosum, have all been reported. There have been ocular abnormalities reported including poor visual development; retinal abnormalities and strabismus. [1-6] To our knowledge there have been no reports in the literature of congenital glaucoma with this chromosome deletion.
Similar to adult onset glaucoma, congenital or infantile glaucoma causes progressive optic neuropathy which can lead to complete blindness if left untreated. Congenital glaucoma is often more severe than primary open angle glaucoma (POAG) due to the timing of the onset of the disease. The ocular structures nor the visual system is fully developed at birth. Interruptions to normal development during this time can lead to irreversible structural and functional damage, including amblyopia. [7]
Congenital glaucoma is said to develop from birth to before the age of three, with the majority of cases presenting prior to six months of age. This type of glaucoma has been classified in varying ways. A simplified classification is divided into primary congenital glaucoma (PCG) and Secondary congenital glaucoma. In primary congenital glaucoma the angle structures are abnormally developed without the presence of other ocular anomalies, causing impaired drainage of the aqueous. Secondary glaucoma is a rise in intraocular pressure caused by other ocular or systemic congenital abnormalities, and glaucoma acquired as a result of ocular disease. Causative agents that have been linked to secondary congenital glaucoma include metabolic, inflammatory, neoplastic, and hamartomatous sources. [7-14] WhilePCG is the cause for 50-70% of all congenital glaucoma’s, it is rare, occurring in approximately 1:10,000 births.The disease is most often bilateral and is found more commonly in males (approximately 65% of all cases). [10,14] About 10% of cases are hereditary and are typically autosomal recessive with variable penetrance. Familial cases have been reported to be demonstrated equally between males and females and occur more frequently in populations with higher prevalence of parental consanguinity. [9,14] In cases of familial inheritance, three major loci have been identified thus far. GLC3A, is by far the most common locus for PCG. This locus is found on chromosome 2p21. Other causative loci been mapped to Chromosome1 and 14. [7,9,10] .
Clinically PCG is characterized by a classic triad of symptoms including epiphora, photophobia and blepharospasm. These signs are not typical of adult onset glaucoma. This difference is due to the elastic characteristics of the infant eye. The collagen fibers of the eye are particularly pliable in the first 3 years of life, thus a rise in intraocular pressure (IOP) can lead to an enlarged cornea (diameter 12mm or greater)(image 1), buphthalmos, Haab’sstriae(linear breaks in Descemet’s membrane), myopia, corneal clouding, and optic neuropathy.[7-14] With the increased IOP, breaks in Descemet’s membrane occur and aqueous can penetrate the corneal stroma and epithelium, causing edema and clouding. The corneal endothelium, which would normally pump aqueous out of the cornea to the anterior chamber, can also be compromised due to the increased IOP, causing further edema. If not treated promptly the corneal edema can lead to permanent scaring (image 2). While corneal clouding will decrease with proper treatment, the Haab’s striae will remain after the IOP is decreased to normal levels. The classic triad of epiphora, photophobia and blepharospasm are induced by the corneal edema, which is very uncomfortable for the infant. [7]
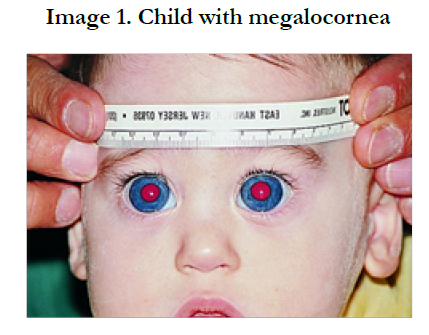
Figure 1. Child with megalocornea [20]
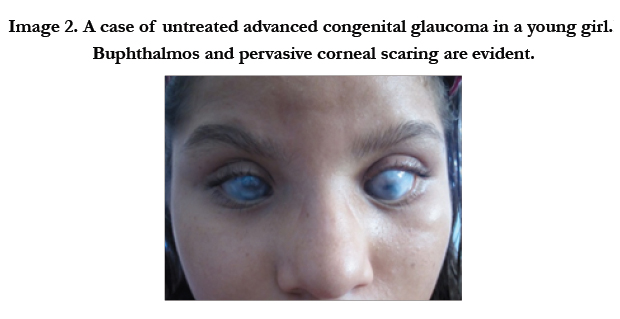
Figure 2. A case of untreated advanced congenital glaucoma in a young girl.Buphthalmos and pervasive corneal scaring are evident.
Given that increased IOP can stretch an infant eye, Myopia is a natural consequence of an elongated eye. Because of the irregular shape of the enlarged cornea, these infants can also demonstrate astigmatism of varying amounts. As with all high myopes, a retinal detachment is often a future concern for children with PCG, even after treatment. [11]
Similar to adult onset glaucoma, a rise in IOP from PCG causes optic neuropathy. However, the optic nerve head cupping seen in PCG is not the sameas cupping due to adult onset glaucoma. PCG occurs at a more rapid pace than that of adult onset glaucoma. In addition, PCG optic nerve head cupping can be reversible if treatment is initiated quickly, prior to irreversible nerve atrophy. The reversible nature of optic nerve head cupping is thought to be due to the pliable nature of the scleral canal, which increases in size with the rise in IOP, causing the optic cup size itself to increase. [7-10] Theearlier a child is treated for PCG, the greater the likelihood that the optic nerve cupping will be reversible. [9]
Severe visual loss from PCG can be reduced with early recognition and treatment. Congenital glaucoma treatment is primarily surgical. [7-14] Surgery, including goniotomy or trabeculotomy, is the treatment of choice in these cases because medical therapy alone has not been shown to lower IOP adequately enough to prevent long term damage from PCG. Medical treatments can also have significant side effects in the infant patient. In addition, surgery for PCG is rather successful and continues to get better, with relatively low complication rates. [7,10,14] Medical therapy can be utilized as an adjunct to surgical treatment. The BIG eye study found that angle surgery combined with medication was the most effective method in lowering IOP in PCG. [14]Prior to surgery, IOP lowing eye drops may help to decrease pain and improve corneal clarity which will improve the surgeon’s view of the anterior chamber angle. [8-10] Pilocarpine is often utilized prior to goniosurgery to open the angle and allow for easier access for the surgeon. Beta blockers have been shown to effectively lower IOP in PCG, however, due to a risk of respiratory and cardio side-effects, they should be used in the lowest dosage available. [8,9,15] Treatment with Prostaglandin analogs have also been shown to be beneficial IOP lowering adjunct therapies in PCG with limited side effects, even when used long-term. [9,16] Due to potentially severe side effects, oral carbonic anhydrase inhibitors and adrenergic agonists are not advised in the medical treatment of PCG. [9]
Case Report
A 19 month old Hispanic male was referred to our clinic by his infant development teacher, who noticed that he was not visually engaged during therapy. The patient was born with a rare chromosome deletion of 6q25. He was delivered at 39 weeks by cesarean section, weighing 6 lbs. 4 oz. His medical history was positive for absence seizures for which he is prescribed Keppra and Phenobarbital. He was severely developmentally delayed in all areas. Despite extensive early intervention therapies, at the time of the exam he had just recently obtained head control and learned to sit without support. He was also very undersize for his age. His ocular history included a previous eye examination at 3 weeks of age. In reviewing those records, he presented with a chief complaint of excessive tearing OU. He was diagnosed with having “blond fundus”, his cup to disc ratios were listed as 0.10/0.10 OU, with all other findings being within normal limits. The plan was to follow up in 6 weeks. Due to dissatisfaction with the previous visit,the parents elected not to return for their 6 week follow up appointment. The patient presented to our clinic about 18 months after that initial evaluation. The parent’s presented with a chief complaint which was shared by the infant development teacher, that the patient was not responsive to visual stimuli. They also explained that the tearing noticed after birth lasted “awhile” but had resolved. They could not estimate how long the tearing lasted and when it ended. They could not recall if photophobia or blepharospasm was exhibited by the patient. Throughout the visit the child was very irritable and difficult to examine. In observing the child’s face, he had buphthalmos OU, which appeared to be pushing on his eyelids. His parents did not recall any changes in the appearance of his eyes during the past 18 months. With our examination we found with Hirschberg testing that his eyes were aligned with no strabismus present. The patient did not demonstrate nystagmus and his eye movements were full and unrestricted OU. A visual acuity measurement could not be obtained with forced preferential looking techniques, however the fix and follow method was utilized. The patient was able to fix and follow with OD and OS. He exhibited an obvious preference for use of his right eye, crying when that eye was covered. He showed no response to the 10 vertical prism test. His pupils were equal and reactive to light, with no APD present. Confrontation field testing indicated restricted visual fields OU, with the left being more restricted than the right. Anterior
segment evaluation indicated megalocornea by appearance and Haab’s striae OS greater than OD.IOP measured by Tono-pen (Medtronic, Jacksonville, Florida) was 18 mmHg OD and 13 mmHg OS.Cycloplegicretinoscopy revealed moderate myopia with astigmatism OU. Upon dilated fundus examination both optic nerves heads appeared larger than average OD and OS, the cup to disc ratios appeared to be approximately 0.80/0.80 OD, 0.95/0.95 OS. His fundus was blond and his macula appeared normal OU. The patient was referred to a pediatric ophthalmologist for a further evaluation. Unfortunately this patient was lost to follow-up; we are unaware of follow up treatment he has received.
Discussion
The patient in our case presents with a deletion of 6q25. In the literature, there are no reported cases of congenital glaucoma in association with this deletion. On the contrary, an over expression of SOD2 gene, mapped to 6q25.3, has been linked to POAG and pseudoexfoliation glaucoma. [17]
In this case, one of the hallmark signs of PCG, very high IOP, was not demonstrated during our examination. However, myopia, Haab’sstriae,and megalocorneaare evidence that the IOP was likely previously elevated. A review of the notes provided from the patient’s exam conducted at 3 weeks of age indicate small cup to disc ratios. At the time of our examination 18 months later, there was extensive optic nerve cupping, worse in the left eye. Even more ominous is the entering complaint of excessive tearing OU at that initial visit when he was 3 weeks old. If in fact this patient was experiencing increased IOP due to PCG during early infancy, prompt and adequate treatment may have prevented the visual dysfunction seen on our examination. There is evidence in the literature of spontaneous resolution of PCG. [18-19] The authors hypothesize that angle structures continue to develop after birth, therefore with the development of a mature angle, the aqueous begins to flow normally and IOP decreases to normal levels. [18-19]
Conclusion
The rare phenomenon of spontaneous resolution of PCG could be the case with the patient presented here. His global developmental delay, growth retardation and dysmorphic facial features caused by his rare 6q25 deletion, may have also delayed the development of his angle causing a period of elevated high IOP, thus causing the glaucomatous-like damage seen on our examination. Due to being lost to follow up, we can confirm that his optic nerve damage remained stable over time. Also without a historical analysis of his angle structures and IOP, we cannot confirm this hypothesis.
References
- Nagamani SCS, Erez A, Eng C. (2009) Interstitial deletion of 6q25.2-q25.3. European Journal of Human Genetics 17: 573-581.
- Michelson M, Ben-Sasson A, Vinkler C. (2012) Delineation of the Interstitial 6q25 Microdeletion Syndrome. American Journal of Medical Genetics Part A 158A:1395-1399.
- Taylor R, Zheng F, Hudson B. (2009) Microdeletion of 6q16.1 encompassing EPHA7 in a child with mild neurological abnormalities and dysmorphic features: case report. Molecular Cytogenetics 2:17.
- Shen-Schwarz S, Hill LM, Surti U, Marchese S. (1989) Deletion of Terminal Portion of 6q: Report of a Case With Unusual Malformations. American Journal of Medical Genetics 32: 81-86.
- Hopkin RJ, Schorry E, Bofinger M. (1997) New Insights into the Phenotypes of 6q Deletions. American Journal of Medical Genetics 70: 377-386.
- Bartoshesky L, Lewis M B, Pashayan HM. (1978) Developmental abnormalities associated with long arm deletion of chromosome No. 6. Clinical Genetics 13: 68-71.
- Kipp MA. (2003) Childhood glaucoma. The Pediatric Clinics of North America 50: 89-104.
- Beck A. (1997) Advances in pediatric glaucoma. Seminars in Ophthalmology 12 (4): 176-189.
- Marchini G, Marco T, Francesca C. (2014) Pediatric glaucoma: current perspectives. Pediatric health, medicine and therapeutics 5: 15-27.
- American Academy of Ophthalmology. Basic and Clinical Science Course. Glaucoma. Section 10. 2011-2012.
- DeLuise VP, Anderson DR. (1983) Primary infantile glaucoma (congenital glaucoma). Survey of Ophthalmology 28 (1): 1-19.
- Hoskins HD, Kass M. (1989) Becker-Shaffer’s Diagnosis and Therapy of the Glaucomas (6th edtn) St. Louis: C.V. Mosby Company. 118–170
- Girgis NM, Frantz KA. (2007) A case of primary congenital glaucoma: A diagnostic dilemma. Optometry 78: 167-175.
- Papadopoulos M, Cable N, Rahi J. (2007) The British Infantile and Childhood Glaucoma (BIG) Eye Study. Investigative Ophthalmology &Vision Science 48(9): 4100-06.
- Plager DA, Whitson JT, Netland PA. (2009) Betaxolol hydrochloride ophthalmic suspension 0.25% and timolol gel-gorming solution 0.25% and 0.5% in pediatric glaucoma: A randomized clinical trial. Journal of American Association for Pediatric Ophthalmology and Strabismus 13(4):384-390
- Uva MG, Avitabile T, Reibaldi M. (2014) Long-term efficacy of latanoprost in primary congenital glaucoma. Eye 28:53-57.
- Lascaratos G, Garway-Heath DF, Willoughby CE. (2012) Mitochondrial dysfunction in glaucoma: Understanding genetic influences. Mitochondrion 12: 202-212.
- Nagao K, Noel LP, Noel ME. (2009) The Spontaneous Resolution of Primary Congenital Glaucoma. Journal of Pediatric Ophthalmology & Strabismus 46 (3): 139-143.
- Lockie P, Elder J. (1989) Spontaneous resolution of primary congenital glaucoma. Australian and New Zealand Journal of Ophthalmology 17 (1): 75-77.
- Olitsky SE, Reynolds JD. (2012) Primary Infantile Glaucoma. Images used with permission. www.uptodate.com





