Pigmentary Retinopathy in Bardet Biedl Syndrome Patient, A Case Report
Reham AlJehani1*, Roaya Ayed Alsulami2, Hala Almotani3, Nawal Alarishi4
1 Ophthalmology Department, King Abdulaziz University, Jeddah, Saudi Arabia.
2 Department of Ophthalmology, East Jeddah Hospital, Jeddah, Saudi Arabia.
3 Department of Ophthalmology, Jeddah Eye Hospital, Jeddah, Saudi Arabia.
4 Department of Ophthalmology, Jeddah Eye Hospital, Jeddah, Saudi Arabia.
*Corresponding Author
Reham AlJehani,
Ophthalmology Department, King Abdulaziz University, Jeddah, Saudi Arabia.
Tel: +966544411428
E-mail: Rehamaljehanii@gmail.com
Received: April 26, 2022; Accepted: May 31, 2022; Published: June 13, 2022
Citation: Reham AlJehani, Roaya Ayed Alsulami, Hala Almotani, Nawal Alarishi. Pigmentary Retinopathy in Bardet Biedl Syndrome Patient, A Case Report. Int J Ophthalmol Eye Res. 2022;10(2):481-483.
Copyright: Reham AlJehani© 2022. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
Abstract
In this case report, we report our approach in a rare case of Bardet Biedl syndrome in 8 years old male patient, found to have poor vision, nystagmus, exotropia, pigmentary retinopathy, polydactyly, truncal obesity, hypogonadism and hypothyroidism. Periodic examination and follow-up are important to prevent disease associated morbidity and mortality due to serious systemic associations. We stress on genetic evaluation especially in consanguineous populations to prevent the recurrence of the disease in future generations.
2.Introduction
3.Materials and Methods
4.Results
5.Discussion
6.Conclusion
7.References
Introduction
Bardet Biedl; Truncal Obesity; Polydactyly; Retinitis Pigmentosa.
Introduction
Bardet-Biedl syndrome (BBS) is a rare ciliopathic autosomal recessive
genetic disorder that affects many organ systems. The
incidence of the syndrome is estimated to be 1:160,000. The
prevalence is much higher in some populations with a high rate
of consanguinity [1, 2]. A previous study published from in Saudi
Arabia on 19 patients representing seven Saudi families, found
a remarkably high frequency of consanguinity in their patients
(100%) compared to the population average (56%)[3]. The main
clinical features of Bardet-Biedl syndrome have been reported as
retinal dystrophy (93%), obesity (91%), cognitive deficit (87%),
hypogonadism (74%), and polydactyly (73%)[4]. Children with
Bardet-Biedl syndrome have a poor visual prognosis with progressive
loss of visual acuity that appears early in the first decade
of life [5]. In this case report, we present and describe our approach
in a case of pigmentary retinopathy due to Bardet-Biedl
syndrome.
Case Presentation
An 8-year-old male patient presented to the clinic with his father
complaining of poor vision since second year of life with nyctalopia
and nystagmus. No other ophthalmological or systemic
symptoms were noted from the parents. He is a product of full
term, healthy pregnancy, spontaneous vaginal delivery with no
complications or neonatal intensive care unit admission. Medically,
he is a known case of hypothyroidism well controlled bythyroxine.
Other review of systems were negative. The family history
revealed epilepsy in one uncle and no cardiac, renal, hematological
diseases. There is positive consanguinity between the parents, but
no relatives were known to have a similar condition or ophthalmological
diseases. Other siblings are normal.
We referred the case to a pediatrician to do full systemic evaluation
and workup. Their evaluation was as follows: The patient
was alert, conscious, active. His vital signs were blood pressure
110/64 mmHg and pulse 110 beats/mins. He is short in stature
with truncal obesity and his weight was 62.3 kg and height
102cm. Facial features showed deep-set eyes and low-set ears. He
has short arms, brachydactyly in hands and polydactyly in his feet.
Respiratory system, cardiovascular, abdominal, and neurological
examination were normal. Examination of genitalia revealed hypogonadism.
Ophthalmological examination showed corrected visual acuity
(VA) of6/60 in the right eye (OD) and counting fingers at 3 meters
in the left eye (OS). Refraction is (OD) -0.50-2.50X10,(OS)
-2.25-2.50X170, orthoptic examination showedExotropia (XT)
of 20prism diopter (PD) for near and distance, Color vision was
completely absent. Extra-ocular muscles movements evaluation
showedfull movements of both eyes with nystagmus which was
jerky, very high frequency, short amplitude, horizontal, uniplanar
and increase in extreme gazes. Pupils were round, regular, reactive.
He has a good red reflex. Lids and lashes were within normal
limits, conjunctiva was quiet, cornea was clear, iris within normal
limits, anterior chamber was deep and quiet. Dilated fundus examination
showed bilateral temporal optic nerve pallor, with cupto-
disc ratio of 0.4,retinal thinning in the macula andpigmentary
retinopathy with attenuated blood vessel. (Figure1) (Figure2).
We wanted to refer the patient for genetic evaluation and his family
for screening, but it was not available in our institute and patient’s
parents were not interested to do that in other institute due
to financial limitations.
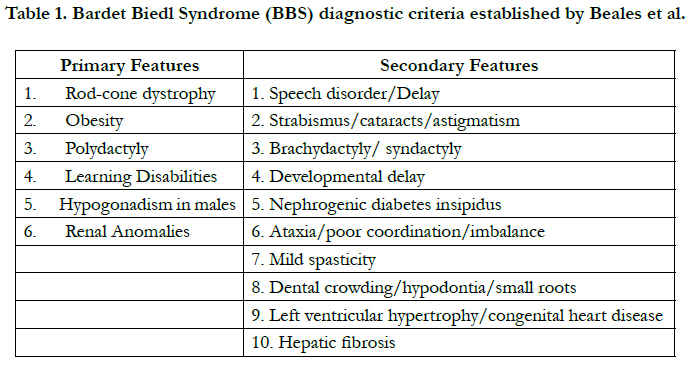

Table 1. Bardet Biedl Syndrome (BBS) diagnostic criteria established by Beales et al.
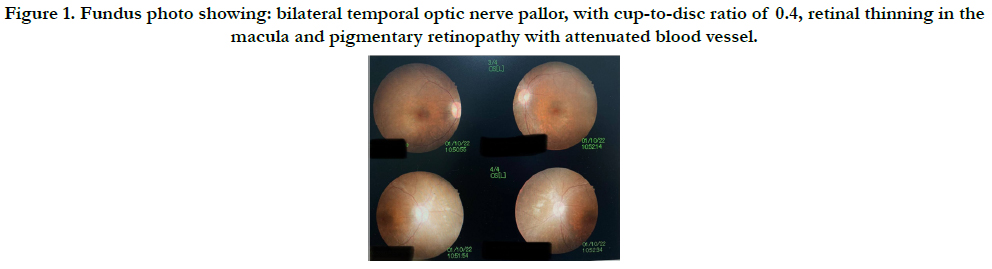

Figure 1. Fundus photo showing: bilateral temporal optic nerve pallor, with cup-to-disc ratio of 0.4, retinal thinning in the macula and pigmentary retinopathy with attenuated blood vessel.
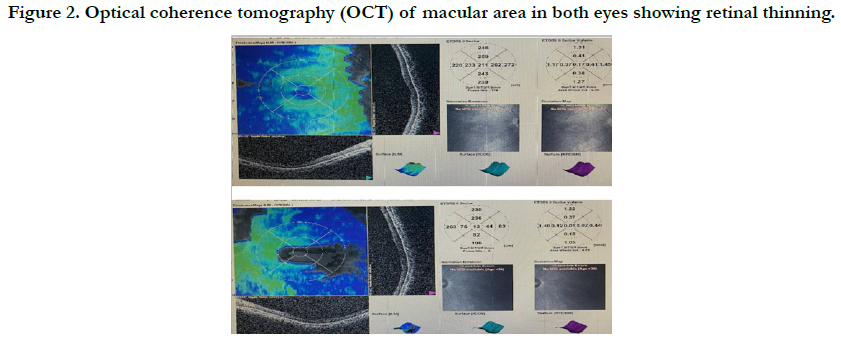

Figure 2. Optical coherence tomography (OCT) of macular area in both eyes showing retinal thinning.
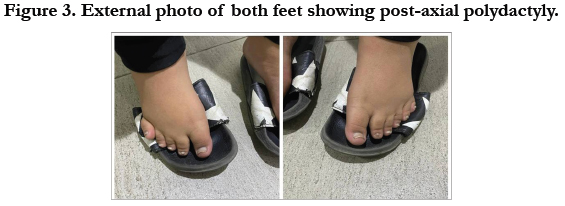

Figure 3. External photo of both feet showing post-axial polydactyly.
Discussion
Bardet-Biedl syndrome (BBS) is a genetic heterogeneous disorder
characterized by a broad spectrum of clinical features described
by Bardet Biedl in 1920 [6, 7]. BBS can result from mutations in
≥20 different genes [1]. Some recent genetic studies that defined
the syndrome by specific genetic mutations. Eleven genes are discovered
to be associated with this syndrome, and those are BBS1,
BBS2, ARL6/BBS3, BBS4, BBS5, MKKS/BBS6, BBS7, TTC8/
BBS8, B1/BBS9, BBS10, TR1M32/BBS11 [8].
Diagnosis is established by clinical findings. Beales et al. have described
the primary and secondary features which are illustrated
in Table (1). The Primary clinical features include retinitis pigmentosa (RP), polydactyly, hypogonadism, obesity, intellectual
disabilities, and renal abnormalities. Secondary clinical features
include speech disorders, strabismus, brachydactyly or syndactyly,
developmental delay, polydipsia-polyuria (diabetes insipidus),
ataxia, spasticity, diabetes mellitus, dental crowding or hypodontia,
congenital heart diseases, and hepatic fibrosis [1]. The presence
of four primary features or three primary features plus two
secondary features is diagnostic [6]. Our patient had four primary
features: Retinitis pigmentosa, Obesity, Polydactyly, and Hypogonadism,
thus fulfilling the diagnostic criteria.
Among all these features, Retinal dystrophy is the most common
and penetrant feature of BBS, making this disorder the second
most common syndromic retinal degeneration behind Usher syndrome
[9, 10]. Retinitis pigmentosa (RP) is the name given to a
group of genetic eye diseases that affect the retina and cause gradual,
permanent loss of vision due to progressive photoreceptor
dysfunction, followed by photoreceptor cell death [5]. RP is the
most common inherited retinal degeneration, with an estimated
worldwide prevalence of 1:4000 [7].
BBS patients typically develop retinitis pigmentosa (RP) symptoms
in the first decade of life and often reach legal blindness
between the second and third decades of life. Night blindness
(nyctalopia) is the most common initial visual symptom and is
usually first noted around 8.5 years [10]. In our patient, the family
start to notice a poor vision at night with nystagmus since birth.
The second prominent feature of BBS is Obesity which usually
begins in childhood. Majority of cases exhibit symptoms within
the first year of life. In our reported case, he developed truncal
obesity in childhood. Hypogonadism is reported more frequently
in BBS males than females [3, 6]. Which is manifested in our patient.
Postaxial polydactyly has been frequently reported in different
ciliopathies, especially in BBS. Our patient has polydactyly in both
hands [11]. Figure (3).
Conclusion
The management of Bardet Biedl Syndrome (BSS) is challenging
and requires multidisciplinary efforts from pediatricians, orthopedic
surgeons, pathologists, audiologists, ophthalmologists, nephrologists,
genetics and other healthcare professionals. Periodic
systemic and ophthalmologic evaluations is the key to prevent the
disease associated morbidity and mortality. Early intervention is
essential in ensuring that children with Bardet Biedl syndrome
reach their highest potential. Genetic evaluation and screening is
helpful especially in consanguineous parents to prevent the recurrence
of similar disease in future off-springs.
References
- Gencol T, Sergin D, Balcioglu T. Anesthetic Management of a Pediatric Patient With Bardet-Biedl Syndrome: A Case Report. A A Pract. 2019 Mar 1;12(5):165-167. PubMed PMID: 30234512.
- Kumar S, Mahajan BB, Mittal J. Bardet-Biedl syndrome: a rare case report from North India. Indian J Dermatol Venereol Leprol. 2012 Mar- Apr;78(2):228. PubMed PMID: 22421669.
- Abu Safieh L, Aldahmesh MA, Shamseldin H, Hashem M, Shaheen R, Alkuraya H, et al. Clinical and molecular characterisation of Bardet-Biedl syndrome in consanguineous populations: the power of homozygosity mapping. J Med Genet. 2010 Apr;47(4):236-41. PubMed PMID: 19858128.
- Hrynchak PK. Bardet-Biedl syndrome. Optom Vis Sci. 2000 May;77(5):236- 43. PubMed PMID: 10831213.
- Andrade LJ, Andrade R, França CS, Bittencourt AV. Pigmentary retinopathy due to Bardet-Biedl syndrome: case report and literature review. Arq Bras Oftalmol. 2009 Sep-Oct;72(5):694-6. PubMed PMID: 20027412.
- Ahmed SN, Shahin MA, Chowdhury R, Ahammad AM, Shazzad MN, Alam MR, et al. A 13-year-old female with Bardet-Biedl syndrome-a case report. Bangladesh J Med. 2015;26(1):31-4.
- Estrada-Cuzcano A, Koenekoop RK, Senechal A, De Baere EB, De Ravel T, Banfi S, et al. BBS1 mutations in a wide spectrum of phenotypes ranging from nonsyndromic retinitis pigmentosa to Bardet-Biedl syndrome. Arch Ophthalmol. 2012 Nov;130(11):1425-32.PubMed PMID: 23143442.
- Khan PA, Nishaat J, Noor S, Fatima N. Laurence-Moon-Bardet-Biedl syndrome: a rare case report in a tertiary care teaching hospital, Hyderabad, Telangana, India. Int J Med Public Health. 2017;7(1):68-71.
- Uğuralp S, Demircan M, Cetin S, Siğirci A. Bardet-Biedl syndrome associated with vaginal atresia: a case report. Turk J Pediatr. 2003 Jul-Sep;45(3):273-5. PubMed PMID: 14696812.
- Weihbrecht K, Goar WA, Pak T, Garrison JE, DeLuca AP, Stone EM, et al. Keeping an Eye on Bardet-Biedl Syndrome: A Comprehensive Review of the Role of Bardet-Biedl Syndrome Genes in the Eye. Med Res Arch. 2017 Sep;5(9). PubMed PMID: 29457131.
- Forsythe E, Beales PL. Bardet-Biedl syndrome. Eur J Hum Genet. 2013 Jan;21(1):8-13. PubMed PMID: 22713813.





