Proniosomes as a Drug Carrier for Transdermal Delivery of Candesartan Cilexetil
Swati Mittal1, Ashu Mittal2, Kiran Sharma2, Sanjar Alam2*
1 Research Scholar, Department of Pharmaceutics KIET School of Pharmacy, Ghaziabad.
2* Department of Pharmaceutics, KIET School of Pharmacy, Ghaziabad, U.P, India
*Corresponding Author
Sanjar Alam
Department of Pharmaceutics, KIET School of Pharmacy,
Ghaziabad, U.P, India
Tel: +91- 9891674226
E-mail: sanjaralam10@gmail.com
Article Type: Research Article
Received: May 08,2013; Accepted: June 22, 2013; Published: June 26, 2013
Citation: Alam S, Mittal S, Mittal A, Sharma K.(2013). Proniosomes as a Drug Carrier for Transdermal Delivery of Candesartan Cilexetil.Int J Nano Stud Technol, 02(02), 17-22. doi: dx.doi.org/10.19070/2167-8685-130004
Copyright: Alam S© 2013. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
Abstract
Candesartan Cilexetil is an angiotensin II receptor antagonist widely used in the treatment of hypertension and characterized by its good efficacy and less side effects compared to other angiotensin II receptor antagonist. The aim of the study was to develop a proniosomal carrier system for Candesartan Cilexetil for the treatment of hypertension that is capable of efficiently delivering entrapped drug over an extended period of time. In the present study transdermal Candesartan Cilexetil proniosomal gels was formulated by using Lecithin, Cholesterol as encapsulating agents, Surfactant, Span and permeation enhancers by coacervation phase separation method. The prepared systems were characterized for pH determination, Viscosity, Vesicle size analysis, rate of spontaneity, encapsulation efficiency, in vitro release study and ex- vivo skin permeation study, skin irritation study and stability study. It was observed that the gel formulations showed good spreadability and viscosity. The particle size was found to be in the range of 175.0- 277.7 nm. The proniosomes showed spherical and homogenous structure in optical microscopy. All formulations showed zero order drug release by diffusion mechanism. The stability studies showed that proniosomal gels were stable at 4 to 8°C and 25±2°C. The above results indicated that the proniosomal gel could be formulated for controlled release of Candesartan Cilexetil. The proniosomal gel are suitable for Candesartan Cilexetil once a day controlled release formulation. The investigated Candesartan cilexetil loaded proniosomal formula proved to be non-irritant, with significantly higher antihypertensive effects and reasonably good stability characteristics.
2.Introduction
3.Materials And Methods
3.1 Preparation of Proniosomes
3.2 Evaluation of Proniosomes
4. Results and Discussion
4.1 pH Determination
4.2 Viscosity Determination
4.3 Entrapment Efficiency
4.4 Optical Microscopy
4.5 Particle size analysis
4.6 Scanning Electron Microscopy (SEM)
4.7 Transmission Electron Microscopy (TEM)
4.8 In vitro release study
4.9 Ex- vivo skin permeation studies
4.10 Skin irritation study
5.Conclusion
6.Declaration
7.Acknowledgements
8.References
Key Words
Proniosomes; Coacervation-Phase Separation; Cholesterol; Lecithin; Controlled Release.
Introduction
Transdermal delivery of drugs through the skin to the systemic circulation provides convenient route of administration for a variety of clinical indications. For transdermal delivery of drugs, stratum corneum is the main barrier layer for permeation of drug. Hence to increase the flux through skin membrane, different approaches of penetration enhancement are used [Hemant etal., 2012]. The main aim of novel drug delivery is to maintain the constant and effective drug level in the body with simultaneous minimization of side effects. Novel drug delivery system also localizes the drug action by placing control release system adjacent to diseased tissue or organ; or target the drug delivery by using drug carriers. At present, not a single drug delivery system fulfills all the criteria but, attempts have been made through novel approaches. Today, numbers of novel approaches have emerged covering various routes of administration, to achieve either controlled or targeted delivery. Vesicular drug delivery is one of the approaches which encapsulates the drug eg. Liposomes, niosomes, transferosomes, pharmacosomes, and provesicles like proliposomes and proniosomes. [Pradnya etal., 2012]
Proniosomes are dry formulations of water-soluble carrier particles that are coated with surfactant and hydrated by agitation in hot water for a short period of time, offer a versatile vesicle delivery concept with the potential for drug delivery via the transdermal route . This would be possible if proniosomes form niosomes following topical application under occlusive conditions, due to hydration by water from the skin itself. The aim of this study is to investigate the feasibility of using proniosomes as a transdermal drug delivery system for Candesartan Cilexetil [Ibrahim etal., 2005]. Vesicles prepared were characterized by optical, scanning and transmission microscopy for vesicle formation and morphology. Drug encapsulation efficiency and release studies were carried out. Finally, a stability study of proniosomal formulations was also performed to investigate the leaking of the drug during storage [ Rishu etal., 2011].
Candesartan Cilexetil proniosomes were prepared by coacervation phase separation method. Precisely weighed amounts of surfactant, lecithin, cholesterol and drug were taken in a clean and dry wide mouthed glass vial of 5.0 ml capacity and alcohol (0.5 ml) was added to it. After warming, all the ingredients were mixed well with a glass rod. The open end of the glass bottle was covered with a lid to prevent the loss of solvent from it and warmed over water bath at 60-70°C for about 5 min until the surfactant mixture was dissolved completely. Then the aqueous phase (0.1% glycerol solution) was added and warmed on a water bath till a clear solution was formed which was converted into proniosomal gel on cooling . [Sanskar etal 2010] (Table 1).
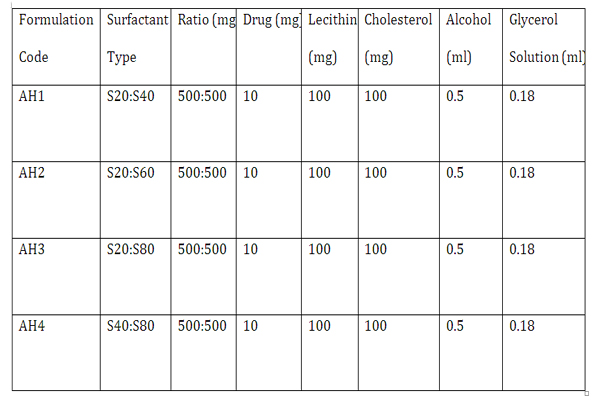

Table 1: Composition of proniosomal formulations prepared.
The pH of each Proniosomal gel was determined using pH meter (Model GMPH, Labindia, Mumbai.). The electrode first calibrated with pH 4.0 and pH 7.0 solution then readings were recorded on pH meter for each formulation (AH1 to AH4). [Hemant etal., 2012]
Viscosities of the formulated Proniosomal gels were determined using Brookfield Viscometer (DV-II + Pro) with T-Shaped spindle set using Spindle no.92 at 2 rpm at 25°C. [Sudhamani etal., 2010].
Viscosities of the formulated Proniosomal gels were determined using Brookfield Viscometer (DV-II + Pro) with T-Shaped spindle set using Spindle no.92 at 2 rpm at 25°C. [Sudhamani etal., 2010].
Percent encapsulation efficiency (EE) as determines by centrifugal method. The proniosomal gel was converted to niosomal dispersion, which as centrifuged (18000 rpm) for 40 min at 5ºC in order to separate unentrapped drug. The supernatant as taken and diluted with phosphate buffer pH 7.4. The drug concentration is the resulting solution was assayed spectrophotometrically at 257 nm. The percentage of drug encapsulation was calculated by the equation .
EE (%) = [(Ct - Cf)/ Ct] × 100
Where Ct is the concentration of total drug and Cf is the concentration of unentrapped drug.[Chandra etal.,2008].
The mean particle size and the polydispersity index (PI) were determined by Malvern Mastersizer (Malvern Instruments Ltd, Malvern, UK). Prior to the measurement, the samples were diluted with double distilled water to suitable scattering intensity and redispersed by handshaking. [Abdallah etal., 2011]
The size and shape of hydrated niosomal dispersion prepared from proniosomes were also determined by SEM. Prior to analysis; the samples were diluted with ultra purified water to obtain a suitable concentration. Then, the samples were spread on a sample holder (stub) and dried using vacuum. They were subsequently subjected to gold palladium coating (JFC 1200 fine coater, JEOL, Japan) and examined by scanning electron microscope (JSM 6301F, JEOL, Japan). SEM, micrograph clearly shows the niosomes of different formulation on the basis of production parameter. [Aqil etal., 2004]
The morphology of hydrated niosomal dispersion prepared from proniosome as also determined by transmission electron microscopy. A drop of niosomal dispersion was applied to a carbon – coated 300- mesh copper grid and left to adhere on the carbon substrate for about 1 min. The remaining dispersion was removed by a piece of filter paper. A drop of 2% aqueous solution of uranyl acetate as applied for 35seconds and again the solution in excess was removed by the tip of filter paper. The sample was air dried and observed under the transmission electron microscope at 90 KV. [Mahmoud etal., 2008]
In vitro release studies on proniosomal gel were performed using locally manufactured Franz-diffusion cell. The area of donor compartment exposed to receptor compartment was 1.41cm2. The dialysis cellophane membrane (MMCO14KDC) was mounted between the donor and receptor compartment. A one gram of proniosomal gel was placed on one side of the dialysis membrane. The receptor medium was containing 200ml solution of isopropyl alcohol and isotonic phosphate buffer of pH 7.4 in the ratio of 20:80. The receptor compartment was surrounded by a water jacket to maintain the temperature at 37±1oC. Heat was provided using a thermostatic hot plate with a magnetic stirrer. The receptor fluid was stirred by a Teflon-coated magnetic bead fitted to a magnetic stirrer. At each sampling interval, samples were withdrawn and were replaced by equal volumes of fresh receptor fluid on each occasion. Samples withdrawn were analyzed spectrophotometrically (Shimadzu-1700) at 257 nm. [Chetna etal., 2011, Narayan etal., 2011]
Permeation of Candesartan Cilexetil, through excised rat skin, from the optimized proniosomal preparation was assessed. The abdominal hair of male wistar rats `( 150 ± 50 gm) was removed carefully. After the animal were sacrificed, the abdominal skin was excised and the adhering fat eliminated. The whole skin was equilibrated in phosphate buffer solution (pH 7.4, the human blood pH) for 1 h before the experiment. [Agaiah etal., 2012] This membrane was mounted on a vertical franz type diffusion cell with the dermis facing the receptor compartment. The donor side was charged with 200 mg of the investigated Preparation. The membrane surface area available for diffusion was 3.14cm2. The receptor compartment was filled with buffer, temperature was maintained at 37 ± 0.50C to simulate human blood temperature. The receptor compartment was constantly stirred at 300 rpm. Sample from the receptor fluid (2ml) were withdrawn at various time intervals upto 24 h and replace immediately by fresh buffer solution; to maintain the “ sink” conditions constantly and a constant volume as well. The samples were then assayed spectrophotometrically at 257 nm.[ Rita etal., 2012]
The albino Wistar rats were housed in polypropylene cages, with free access to standard laboratory diet and water. Animals were acclimatized for at least 7 days before experimentation. The dorsal abdominal skin of rats was shaved 24 h before study. Transdermal gel was applied and site of application was occluded with gauze and covered with a nonsensitizing microporous tapes. Formalin (0.8 %w/v) solution was applied as standard skin irritant. The formulation was removed after 24 h and score of erythema was recorded and was compared with standard.[ Raja etal., 2011]
Stability studies were carried out by storing the optimized formulation (AH2) at various temperature conditions as per ICH guidelines i.e. at refrigeration temperature (2°-8°C), room temperature (25°± 0.5°C) for a period of two months. Entrapment Efficiency and variation in the average vesicle diameter were determined before and after the completion of 2 months.[Kandasamy etal., 2010]
All data are reported as mean ± standard error of mean and the differences between the groups were tested using Student’s t-test at a significance level of P< 0.05. More than two groups were compared using analysis of variance and the difference of P < 0.05 was considered significant.
Skin compatibility is the primary requirement for a good topical formulation, it was found that the pH of all the formulations were in the range of 5.24 to 7.40 that suits the skin pH, indicating skin compatibility. The results of pH determination are reported in Table2.
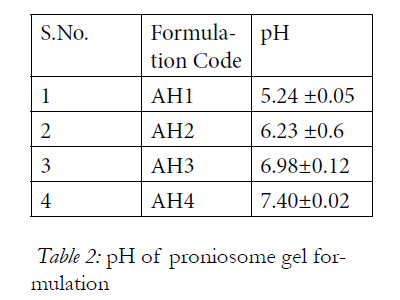
Table 2: pH of proniosome gel formulation.
Viscosity measurement of all the formulations revealed optimum consistency and the results are reported in Table 3
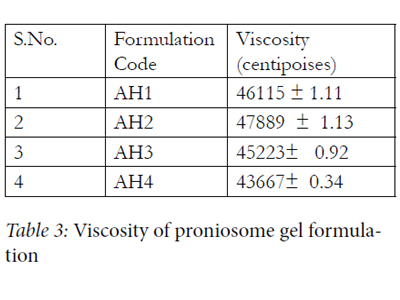
Table 3: Viscosity of proniosome gel formulation.
The EE of all proniosomal formulations are reported in Table 4. Candesartan Cilexetil was best encapsulated by proniosomal prepared using Span 20 : Span60 (AH2). This might be attributed to the fact that Spans 60 and 40 are solid at room temperature and showed a higher phase transition temperatures [Tc]. It was found from the result that the entrapment efficiency of Span 80 formulations was less than those of Span 60 formulations as Spans 60 and 80 have the same head group, but Span 80 has an unsaturated alkyl chain. Introduction of a double bond into the paraffin chain causes a marked enhancement in the permeability in liposomes. This might be the reason for the lower entrapment efficiency of the Span 80 system.
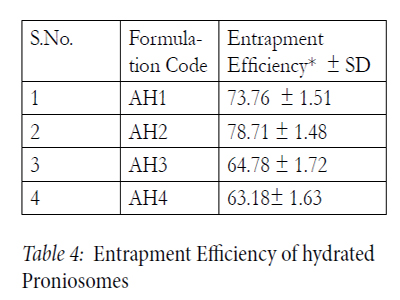
Table 4: Entrapment Efficiency of hydrated Proniosomes.
The photomicrographs of hydrated AH2 optimized proniosomal formulations, composed of Span 20 : Span60 respectively. The prepared vesicles were studied under 400x magnifications to observe the formation of vesicles. Some unevenness of vesicles that observed under the study may be due to drying process under normal environment condition. The photographs revealed that the niosomes are unilamellar vesicles having spherical shape and no aggregation or agglomeration is observed.
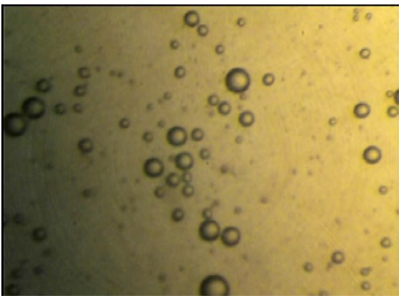

Figure 1: Photomicrograph of hydrated AH2 Proniosomal formulation.
The particle size and polydispersity index (PDI) were determined by Zetasizer Nano ZS (Malvern Instruments Ltd, Malvern, UK). The particles exhibit Brownian motion, which causes the intensity of light to scatter from particles, which is then detected as a change in inten¬sity with suitable optics and a photo multiplier. All the data analyses were performed in automatic mode with triplicate measurement within each run. Mean Average particle size of formulations AH1, AH2, AH3 and AH4 were found to be 277.8, 227.5, 203.1 and 175.6 respectively. Polydispersity index of these formulations were found to be 0.900, 0.270, 0.311 and 0.303 respectively in Table5.
Determination of particle size is an important parameter for the topical application. Size was reduced when the dispersion was agitated. The reason for this is the energy applied in the agitation which results in the breakage of the larger vesicles to smaller vesicles. The size range was found to be 175.6-277.8 nm. Particle size of optimized AH2 formulation was found to be 227.5nm. Particle size was found to be smallest in AH4 formulation (175.6 nm) due to the presence of Span 80. Increasing hydrophobicity of the surfactant monomer led to a smaller vesicle, a result that is expected since surface energy decreases with increasing hydrophobicity.<
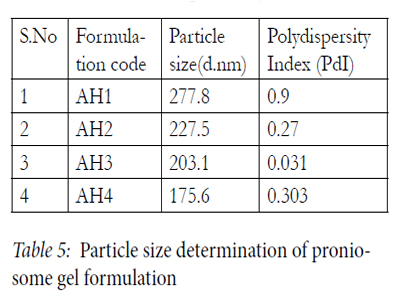
Table 5: Particle size determination of proniosome gel formulation.
Surface morphology confirms the coating of surfactant in carrier. Results of scanning electron microscopic study of niosomes prepared from AH2 proniosomal formulations shows the vesicles are well identified, spherical and discreet with sharp boundaries having large internal aqueous space.
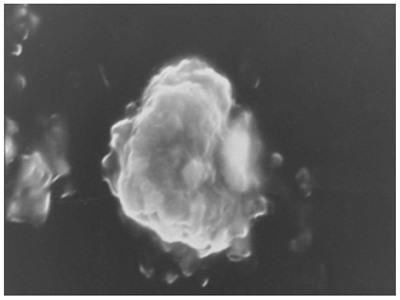
Figure 2: Scanning electron image of hydrated AH2proniosomal formulation(at 25 kV with magnification 2,000 X).
Results of transmission electron microscopic study of niosomes prepared from AH2 proniosomal formulations shows the vesicles are well identified, spherical and discreet with sharp boundaries having large internal aqueous space.
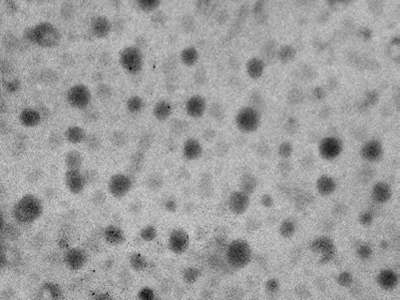

Figure 3: Transmission electron micrograph of hydrated AH2 proniosomal formulation (at 90 kV with magnification 100,000X).
The release of Candesartan Cilexetil from the investigated stable formulation follows zero order kinetics with an immediate release of Candesartan Cilexetil (no lag time). Comparing the release profile of other formulation it was found that the formulation which have increasing hydrophobicity shows better release. Results are shown in Figure. 4.
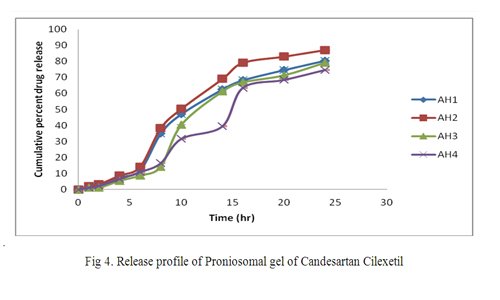

Figure 4: Release profile of Proniosomal gel of Candesartan Cilexetil.
Figure 5 shows that permeation of Candesartan Cilexetil entrapped in proniosomal optimized formulation through the skin follows zero order kinetics. It is also evident that the optimized formulation effectively enhanced Candesartan Cilexetil permeation, showing higher flux value. In order to verify the predominant driving force for Candesartan Cilexetil proniosomes permeating across skin, some efforts were made to clarify these mechanisms. The release rate of the drug across cellophane membrane was previously determined and compared to its flux through rat skin.
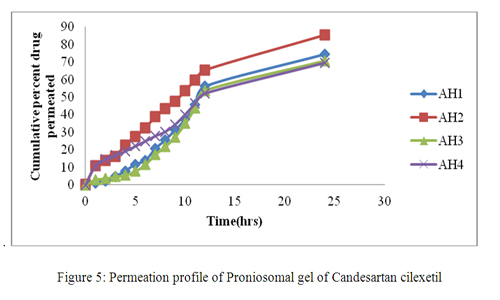
Figure 5: Permeation profile of Proniosomal gel of Candesartan cilexetil.
No erythema was found after 7 days when optimized formulation AH2was applied to the dorsal skin of rat and compared with standard irritant. Thus the formulation was found to be suitable for transdermal application.
From the results of vesicular size and shape and entrapment efficiency of the optimized formulation (AH2) it was concluded that the formulation was stable at refrigeration and room temperature as well (Table 6)
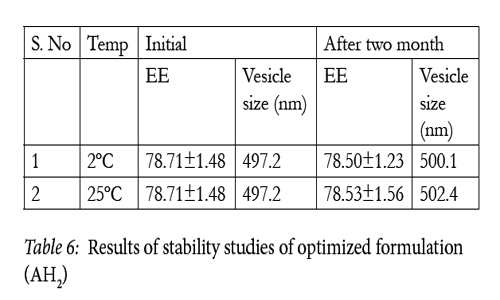
Table 6: Results of stability studies of optimized formulation (AH2).
Conclusion
The Transdermal Proniosomal Gels showed controlled drug release properties. The results of the present study indicated that Candesartan Cilexetil proniosomal gel containing lecithin, cholesterol and in combination of surfactants like span 20, 40, 60, 80 shows sustained release of drug over a period of 24 hrs for the management of hypertension. The optimized AH2 proniosomal gel system showed better in-vitro and ex-vivo permeation study and will be a great potential for delivery of anti hypertensive drug Candesartan Cilexetil. Thus proniosomal gel could be an effective alternative vehicle for delivering the drug through transdermal route to avoid side effects associated with oral route. Thus Proniosomes may be a promising carrier for Candesartan Cilexetil and other drugs, especially due to their simple production and facile scale up. However, benefit-to-risk ratio and clinical intricacies need to be established scientifically through in vivo studies, for its suitability in clinical practice in the monitoring of antihypertensive activity of the prepared proniosomal gel through topical route. Thus, in conclusion, proniosomal formulation of Candesartan Cilexetil holds an immense potential for the development of topical antihypertensive agent comparable to conventional oral antihypertensive agent.
Declaration
The author(s) declare that they have no competing interests or financial benefit from this work.
Acknowledgements
Authors are thankful to the principal KIET school of Pharmacy Ghaziabad for Providing the best research facilities available for conducting the research work. Authors are also thankful for the support provided by Advanced Instrumentation Research Facility (AIRF), Jawaharlal Nehru University and Nano Medicine lab, Faculty of Pharmacy, Jamia Hamdard, New Delhi.
References
- Hemant, N. P., Sharwaree, R., Hardikar, and Ashok, V.B. Int. J. Pharm 2012, 1: 191-197.
- Pradnya, C., Bharat, J., and Parag, J. (2012) World J. of Pharmacy and Pharmaceutical sci 2012, 1 : 393-404.
- Ibrahim, A. A., Bosela, A.A., Ahmed, S.M., and Mahrous, G.M. European J. ofpharmaceutics and Biopham 2005, 59: 485–490.
- Rishu, K., Rekha, R., Navin, K.D., and Sanju, N. Majeo Int. J. of Sci. and Technology 2011, 01: 146-158.
- Ankur, G., Sunil, K. P., Mamta, B., S., and Daksh B. Tropical J. of Pharmaceutical Research 2007, 2: 687-693.
- Shamsheer, A. S., Sabareesh, M., Rafi, K., Sai, k. P., and Sudheer B. J.of Applied Pharmaceutical Sci 2011, 08: 181-185.
- Chandra, A., and Pavan, K. S. African J. of Pharmacy and pharmacology 2008, 9: 184-190.
- Abdallah, M., Sammour, O., Ghamry, H., and Nahas H. Int. J. Pharm 2011, 8: 2195-2205.
- Mahmoud, M., Omaima, A. S., Mohammed, A. H., and Nagia,A. M. Int. J. Pharm 2008, 1: 104–111.
- Chetna, G., Munish, A., and Surendra, K. S. Acta Poloniae Pharmaceutica - Drug Research 2011, 1: 147- 150.
- Rita, B., and Lakshmi, P.K. J. of Applied Pharmaceutical Sci 2012, 3: 85-91.
- Raja, K., Jestin, P. U., Athul, P. V., Tamizharasi, S., and Sivakumar. T. Int. J. Pharm 2011, 3: 471-477.
- Aqil, M., Ali, A., and Sultana, Y. Ethiop. J. Pharm 2004, 22:53–60.
- Agaiah, G., Raju, J., and Rambhau, D. Int. J. Pharm 2012, 1: 37-42.
- Kandasamy, R., and Veintramuthu, S. AaAPS Pharma Sci. Tech 2010, 3: 134-167.
- Sudhamani, T., Priyadarisini, N., and Radhakrishnan, M. Int.J. Pharm 2010, 1: 1446-1454.
- Sankar, V., Ruckmani, K., Durga, S., and Jailani, S. Pak. J. Pharm. Sci 2010, 1: 103-107.
- Narayan, D. S., and Manika, T. Int. J. of Research in Pharmaceuticaland Biomedical Sci 2011., 3: 880-887.





