Nanoscale Small Interfering RNA Delivery Systems For Personalized Cancer Therapy
Fatih Uckun*1,2 & Seang Yiv 1
*1 Developmental Therapeutics Program, Children’s Center for Cancer and Blood Diseases, Children’s Hospital Los Angeles MS#160, Los Angeles, CA 90027, USA. Email Id: fmuckun@chla.usc.edu
2 Division of Hematology-Oncology, Department of Pediatrics, Children’s Center for Cancer and Blood Diseases, University of Southern California Keck School of Medicine and, Los Angeles, CA 90027-0367,USA.
*Corresponding Author
Fatih Uckun,
Developmental Therapeutics Program,
Children’s Center for Cancer and Blood Diseases,
Children’s Hospital Los Angeles,
MS#160, Los Angeles, CA 90027, USA.
Email: fmuckun@chla.usc.edu
Article Type: Research Article
Received: June 10, 2012; Accepted: August 16, 2012; Published: August 22, 2012
Citation: Uckun F, Yiv S (2012). Nanoscale Small Interfering RNA DeliverySystems For Personalized Cancer Therapy, Int J Nano Stud Technol, 1(2), 6-11. doi: dx.doi.org/10.19070/2167-8685-120002
Copyright: Fatih Uckun© 2012 This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
Abstract
Nanoparticles represent particularly attractive delivery systems for small interfering RNA (siRNA) and may provide the foundation for rational design and formulation of RNAi-triggering nanomedicines. siRNA can be delivered with a therapeutic intent using lipid-based delivery platforms such as stable nucleic acid lipid particles (SNALP) with a lipid bilayer containing cationic as well as fusogenic lipids and a diffusible PEG-lipid coat, polymers, cationic complexes, recombinant fusion proteins, conjugates, or polyconjugates. Several investigators have reported preclinical or early clinical proof of concept studies demonstrating that systemic delivery of siRNA nanoparticles targeting specific gene transcripts can elicit biologic responses. Therapeutic nanoparticles containing siRNA targeting specific genes that contribute to the aggressiveness and/or radiochemotherapy resistance of cancer cells may facilitate a paradigm shift in modern cancer therapy.
2.Introduction
3.Strategies for Packaging siRNA into Liposomal Nanoparticles
4. PEGylation of siRNA Nanoparticles
5. Renal Disassembly and Clearance of siRNANanoparticles
6.Target Disease Considerations for siRNA Nanoparticles
7.Intracellular Trafficking of siRNAnanoparticles
8.Multifunctional Targeted siRNA Nanoparticles Against Leukemias and Lymphomas
9.References
Key Words
Personalized medicine, Cancer, Nanomedicine, Biotherapy, Leukemia, siRNA
Introduction
RNA interference (RNAi) has emerged as an attractive technology for silencing the expression of specific genes in human cells [1-6]. In the physiological RNA interference pathway of gene silencing, double stranded RNAs are processed into small interfering RNAs (siRNA) by the RNase enzyme DICER. These siRNAs are incorporated into an RNA-induced silencing complex (RISC), that is capable of identifying and degrading mRNA that is complementary to the antisense strand of the siRNA thereby causing “gene silencing” [1-6]. However, sequence-specific gene knockdown via RNAi can also be triggered by a variety of synthetic double-stranded siRNA species that are capable of serving as DICER substrates and are therefore being developed as potential RNAi therapeutics candidates [1-6]. Several formidable obstacles exist for the development of siRNA as RNAi therapeutics, including their rapid degradation by nucleases in the blood, poor cellular uptake, and requirements for endosomal escape after cellular uptake, off-target effects due to their microRNA-like activity profile, and their inflammatory effects [7-9]. It remains to be seen if specific formulation strategies or structural modifications in the synthetic siRNA molecules can effectively overcome these obstacles or prevent inflammatory acute immune responses, including activation of innate immune receptors and/or the complement system and release of proinflammatory cytokines.
Nanoparticles represent particularly attractive delivery systems for siRNA and may provide the foundation for rational design and formulation of RNAi-triggering nanomedicines. The siRNA delivery systems that have emerged during the last decade revolve around liposomal formulations [1-10], non-bilayer self-assembled polymeric nanoparticles [9, 11-15], and to a lesser extent, tertiary hybrid systems where lipids, polymers or solid cores are utilized [16-18]. The pharmacological effectiveness of oligonucleotide-based therapeutics depends on their cellular uptake, intracelular trafficking, endosomal release, and productive delivery to their target subcellular compartments [19,20]. The uptake of siRNA into the target cells is possible only if their highly polyanionic charges are hidden from the hydrophobic cell membrane bilayers. This may be achieved by presenting siRNA to the cells in a condensed form where the siRNA charges are neutralized by cationic materials. The condensation of the siRNA can be enabled by the formation of polyanion-cation complex, arising from the non-covalent electrostatic interaction between the cationic charges embedded in the nanoparticles and the polyanionic siRNA. In nearly all of the contemporary siRNA systemic delivery systems in development, the use of siRNA complexation has become almost a universal procedure for packaging siRNA into either lipid-based or polymer-based nanoparticles. siRNA cargo is complexed and compressed, meaning that its polyanionic charges are neutralized and its size made smaller [1-18]. Compression of the siRNA cargo takes place when it forms a complex with cationic materials including cationic lipids (lipoplex) and polycationic polymers (polyplex). The design of siRNA packaging has progressed in both directions, using cationic liposomal and polymeric nanoparticles [2-15] although at least ten siRNA lipid systems [3, 21-24] - compared to only one siRNA polymeric nanoparticle [11,13] - have reached early clinical stages in development.
Following cellular uptake, generally through endocytosis, the siRNA-carrying nanoparticles and/or their cargo must escape the endosomes and enter the cytoplasm [19, 20]. While the packaging of siRNA into nanoparticles via complexation promotes its delivery into the target cells, siRNA must eventually be released from the nanoparticles into the cytoplasm in order to trigger RNAi. siRNA can be delivered with a therapeutic intent using lipid-based delivery platforms such as stable nucleic acid lipid particles (SNALP)
with a lipid bilayer containing cationic as well as fusogenic lipids and a diffusible PEG-lipid coat, polymers, cationic complexes, recombinant fusion proteins, conjugates, or polyconjugates [1-3, 7-9, 19, 20, 25-34]. Several investigators have reported preclinical and early clinical proof of concept studies demonstrating that systemic delivery of an siRNA nanoparticle targeting a specific gene transcript can elicit biologic responses in vivo [11, 35-42]. Davis et al. reported siRNA-loaded multifunctional nanoparticles that consist of a cyclodextrin-based synthetic polymer, a transferring receptor ligand for active targeting, and polyethylene glycol as a hydrophilic polymer for nanoparticle stability [11]. Afonin et al. developed self-assembling functional nanoparticles for siRNA delivery using two complementary nanoscaffold designs (nanoring and nanocube), which serve as carriers of multiple siRNAs [35]. Lee et al. recently reported the synthesis of RNAi-microsponges as a novel nanoscale delivery vehicle in which RNAi polymers that self-assemble into nanoscale pleated sheets of hairpin RNA, which in turn form sponge-like microspheres. The RNAi-microsponges are processed to siRNA only after cellular uptake [36]. Sakurai et al reported the use of a fusogenic peptide to modify liposomes to enhance the endosomal escape of the encapsulated siRNA delivered as the payload against cancer cells (37). Polymeric vectors such as polyethylenimine (PEI) have also been used for siRNA delivery [38]. Liu et al. reported the use of an amphiphilic block copolymer composed of conventional monomethoxy (polyethylene glycol)-poly (d, l-lactide-co-glycolide)-poly (l-lysine) (mPEG-PLGA-b-PLL) to effectively deliver siRNA to cancer cells both in vitro and in vivo [39]. Others have employed nanodiamonds that are coated with a cationic polymer for siRNA delivery [40]. Sparks et al. reported a class of structurally versatile cationic lipopolyamines including staramine as a core lipid designed specifically for effective delivery of siRNA [41].
In recent years, several recombinant fusion proteins that consist of a cell surface targeting moiety (e.g an antibody fragment or ligand) and an oligonucleotide complexation moiety (e.g truncated protamine) have been designed for targeted delivery of siRNA [27-29, 42]. The oligonucleotide complexing cationic moieties
condense and mask the negative charge of the oligonucleotides and thereby assist their uptake through the cell membrane. Furthermore, they are capable of rupturing endosomes by a proton-sponge effect and promote the release of siRNA into the cytoplasm [29].
These nanoscale delivery platforms offer several theoretical and practical advantages over more traditional lipid-based nanoparticle formulations [29]. Song et al. reported the preclinical proof of concept that such fusion proteins can selectively deliver effective doses of siRNA to cancer cells both in vitro and in vivo [28]. Further preclinical and clinical development of the most promising nanoscale delivery platforms for selective delivery of siRNA may provide the foundation for much needed therapeutic innovations against various forms of cancer, especially those that do not respond to contemporary chemotherapy or radiation therapy regimens.
Strategies for Packaging siRNA into Liposomal Nanoparticles
Figure 1 shows various cationic and neutral/helper surfactants that have been used in the liposomal siRNA nanoparticle formulations. Early studies have emphasized the use of a mixture of cationic lipids, such as 2, 3-dioleyloxypropyltrimethylammonium chloride (DOTAP), with a helper neutral lipid, such as 1, 2-Dioleoyl-sn-Glycero-3-Phosphoethanolamine (DOPE). But in recent years, the availability of rationally designed cationic lipids, such as Lipidoid and SNALP, has allowed the design and engineering of increasingly more sophisticated nanoscale formulations for more effective siRNA delivery [31-34]. The complexation of siRNA in the liposomal nanoparticles has been commonly driven by strong electrostatic interactions between polyanionic siRNA and cationic lipids, when preformed liposomes and a siRNA in a buffer solution are mixed together. Particle size reduction to a desirable particle size range (generally below 120 nm) can be achieved by using high shear homogenization and extrusion before mixing with siRNA [31-34]. In an effort aimed at reducing the toxicity associated with use of cationic lipids, some investigators have employed neutral lipids in the preparation of liposomal siRNA nanoparticles [21-23]. Alternatively, nanoparticles can be prepared without an extrusion step under conditions where the lipid concentrations fall below their solubilities in the mixed solvents, leading to formation of liposomes with particle diameter <200 nm without any needs of extrusion [43]. In addition, some investigators have reported preparation of siRNA-loaded liposomes by using a freeze-dry procedure [45], although this procedure may result in unintended structural alterations [46]. Moreover, to package siRNA, some groups utilized significantly more complex liposome formulations. For example, in addition to a cationic lipid and a neutral or helper lipid in the formulations, cationic polymers (protamine) and DNA (or hyaluronic acid) have been employed to create a virus-like core [16]. Calcium phosphate [17] or calcium carbonate [18] in solid states, which dissolve at low pH in the endosomes, have also been utilized as the cationic core of the liposomes. It remains to be seen which of these various alternative formulation strategies will prove most practical and cost-effective.
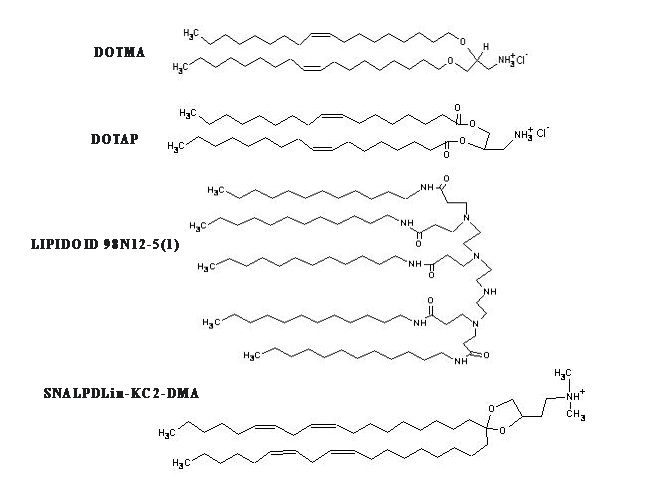
Figure 1: Structures of cationic surfactants used in various siRNA liposomal delivery nanoparticles.
DOTMA: 1, 2-di-O-octadecenyl-3-trimethylammonium propane (chloride salt);
DOTAP: 2, 3-dioleyloxypropyltrimethylammonium chloride; LIPIDOID 98N12-5(1) [34];
SNALP Dlin-KC2-DMA [37]; MVL5 [49]; Atu01 [45];
DOSPA: 2,3-dioleyloxy-N- [2(sperminecarboxamido)ethyl]-N,N-dimethyl-1-propanaminium trifluoroacetate
PEGylation of siRNA Nanoparticle
Nanoparticles have been coated with polyethylene glycol (PEG) (i.e., PEGylated) in an attempt to render them resistant against protein adsorption, enhance their biocompatibility, and to stabilize them against agglomeration in biological environments. PEGylated nanoparticles with diameters around 100 nm may become long-circulating in the blood stream and have been called stealth particles since they can evade recognition by T cells and macrophages and avoid rapid clearance by the immune system [47-54]. Nanoparticles that are sterically stabilized by PEG polymers on their surface and have surface charges that are slightly negative or slightly positive have minimal self-self or self-non-self interactions and improved pharmacokinetics [51-54]. PEGylation of nanoparticles creates a hydrophilic surface and leads to increased protein solubility, reduced immunogenicity, prolonged plasma half life due to prevention of rapid renal clearance, and reduced clearance by the RES system due to decreased macrophage capture and opsonization [51-54]. The vast majority of the siRNA nanoparticles in preclinical or clinical development are PEGylated. However, PEGylation must be optimized as the hydrophilic PEG molecules may negatively influence the cellular uptake of nanoparticles through the lipophilic cellular membrane.
Renal Disassembly and Clearance of siRNANanoparticles
A unique pharmacologic problem, namely premature disassembly and renal clearance, has recently been discovered for polymeric siRNA nanoparticles [55]. The nature of the siRNA-cationic polymer complex and the zeta potential of the nanoparticles have been shown to play a critical role in their potential clearance by the glomerular basement membrane (GBM) [55]. Lipoplex nanoparticles, with a size range of 100 nm or smaller, a positive zeta potential, and held together mainly by electrostatic interaction, may be susceptible to disassembly by the abundance of heparin sulfate in the GBM. For most liposomes, siRNA molecules are encapsulated and separated by at least one lipid bilayer as well as by a PEG-shielding from the external phase. Therefore, a GBM-mediated disassembly would be less likely albeit still possible, depending on the strength of the electrostatic interaction of the specific cationic lipid used and siRNA compared to that of between heparin sulfate and siRNA. In view of the observed GBM-associated disassembly and rapid renal clearance of polymeric siRNA nanoparticles, it is imperative that product release criteria for such nanoparticles be revised to include measures of the strength of the siRNA-cationic lipid complex in the nanoparticles. Developers should carefully select their cationic materials and optimize the nanoparticle formulation to minimize the risk of disassembly and incapacitation by heparin sulfate in the GBM causing premature clearance through the kidney.
Target Disease Considerations for siRNA Nanoparticles
Nanotechnology-enabled delivery of anti-cancer therapeutics is an area of intense translational research [47-56]. Rationally designed biotargeted anti-cancer nanomedicines have the potential to substantially improve the therapeutic index of their “payload” by increasing their potency via (a) selective delivery to target cancer cells as well as (b) improved cellular pharmacokinetic/pharmacodynamics (PK/PD) features that avoid the multi-drug resistance associated drug efflux pumps and by reducing their systemic toxicity and undesired off target effects. Several non-targeted nanomedicine candidates are being evaluated in clinical trials or have been given FDA approval, including biocompatible micellar, liposomal, and polymeric formulations of standard chemotherapy drugs. Several biotargeting moieties are being explored in pre-clinical studies including small molecules, antibodies/antibody fragments, affibodies, cell penetrating peptides, cytokines, avimers and aptamers. Current research efforts in several laboratories are aimed at overcoming the barriers that limit the effective tumor delivery and penetration of the nanomedicine candidates as anti-cancer therapeutics, including: [a] heterogeneous tumor circulation caused by abnormal and irregular architecture of the tumor vasculature, [b] intratumoral vascular hyperpermeability contributing to increased interstitial pressure in the targeted tumor that substantially reduces the convective transport of nanoparticles and [c] impaired diffusion in the context of an abnormal and highly dense extracellular collagen matrix in the tumor microenvironment. Unlike nanoparticles carrying cytotoxic small molecule chemical compounds that can leak out of the nanoparticles and reach target sites via diffusion, siRNA nanoparticles must reach and taken up by each target cancer cell in order to be effective [19, 20]. Therefore, solid tumors are likely to be more resistant to siRNA nanoparticles than hematologic malignancies that do not have the physical barriers associated with solid tumors [57,58].
Intracellular Trafficking of siRNAnanoparticles
Following their cellular uptake, siRNA nanoparticles and/or their cargo must escape endosomes in order to reach their target sites. If the siRNA-carrying nanoparticles are compartmentalized too long in the endolysosomal vesicles, siRNA would be degraded by nucleases of the acidic and nuclease-rich endolysosomal microenvironment. In order to prevent their premature degradation, siRNA nanoparticles must be rationally designed and engineered [19, 20]. The schematic in Figure 2 describes the endosomal escape of liposomes through the fusion or intermixing of nanoparticle bilayers and endosomal membrane. Before the exposure to low endosomal pH, the liposomal as well as endosomal lipids are in a headgroup (H) to tail (T) balance favoring the formation of lamellar layers (nearly equal T and H surface areas). But ion-pairing at the endosomal low pH induces a sharp drop in the surface area of the ion-pair combined headgroup and a spreading of the hydrophobic tails. The membrane destabilization triggered by ion-pairing needs to be sufficiently strong and rapid for a timely escape and possibly some partial or complete siRNA-cationic lipid decomplexation. The break-up of the labile T-T, T-H, or H-H linkers would play a supporting role in assuring a more complete process. According to the equations in Figure 3, high fusogenic potency (FP), which represents the strength of the destabilization and fusion process, can be obtained directly by modifying all parts (H, linkers, and T) of the cationic lipids. Cationic lipids with largest FP can be obtained by designing a lipid molecule which assumes a nearly equal lipid tail to headgroup surface area ratio (ACH ~ ACL) at physiological pH but a highest ratio of lipid tail to ion-pair surface area (ATL/ATH) at endosomal pH (Equation 3, Figure 3). The membrane destabilization and rupture can be achieved by designing nanoparticles containing bilayer materials capable of effectively destabilizing and rupturing the endolysosomal membranes at the endosomal pH (Figure 3). As illustrated in Figure 3, the sharp and rapid decrease of the surface area of the combined headgroups, triggered by the ion-pairing between liposomal cationic lipids and endosomal anionic lipids, causes the surface curvature to be much more favorable for a non-bilayer hexagonal phase (HII) or inverted micelles. This inversion in spontaneous curvature in the context of the lower pH of the endosomal environment must be sufficiently large, for it to disturb and burst open the endosomal membrane and release the siRNA-containing nanoparticles into the cytosol.
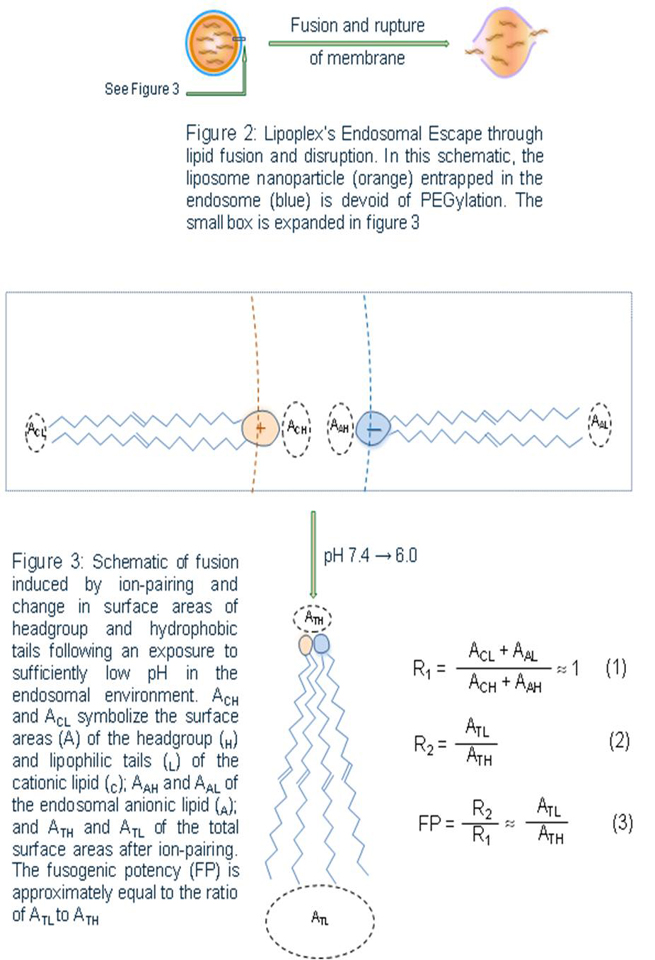

Figure 1: Structures of cationic surfactants used in various siRNA liposomal delivery nanoparticles.
Multifunctional Targeted siRNA Nanoparticles Against Leukemias and Lymphomas
The pharmacological effectiveness of siRNA-based therapeutics depends on their cellular uptake, intracelular trafficking, endosomal release, and productive delivery to their target subcellular compartments [19, 20]. siRNA Nanoparticles can be functionalized with a tumor targeting moiety such as a ligand or scFv directed against a surface receptor on cancer cells in order to achieve optimal tumor targeting and site-specific drug delivery to further reduce their toxicity and improve their efficacy [51, 54]. When linked with tumor targeting moieties nanoparticles can reach cancer cells carrying the target receptors with high affinity and precision [51-54]. The targeting ligands enable nanoparticles to bind to cell surface receptors and enter cells by receptor-mediated endocytosis [19]. An excellent potential target for the biotargeting moieties of siRNA nanoparticles is the CD19 receptor of B-lineage leukemia/lymphoma cells [59-61].
CD19 is a 95-kDa B-lineage restricted receptor molecule that functions as a key regulator of transmembrane signals in both B-cells and B-cell precursors [59-61]. CD19 antigen is acquired at a very early stage of B-cell ontogeny, prior to rearrangement of immunoglobulin genes and expression of other B-precursor antigens such as CD10 and CD22 (59-61]. CD19 antigen is abundantly expressed on the malignant cells from B-lineage leukemia and lymphoma patients, but it is absent on the parenchymal cells of life-maintaining non-hematopoietic organs, circulating blood myeloid and erythroid cells, T-cells as well as bone marrow stem cells, [59-61]. In B-lineage ALL, CD19 antigen is expressed on candidate leukemic stem cell populations with in vivo clonogenic, leukemia initiating and propagating properties in xenograft models using immunocompromised mice. The very favorable B-lineage leukemia/lymphoma vs. normal tissue expression profile of CD19 and its association with stemness properties of leukemic B-cell precursor populations make it an attractive molecular target for biotherapy in relapsed ALL [59-61]. Several hundred thousand CD19 molecules located on the surface of each B-lineage leukemia/lymphoma cell are rapidly internalized upon ligation with anti-CD19 mAb or immunoconjugates (59, 62). CD19 has tyrosine-based internalization motifs in its cytoplasmic domain that were predicted to bind the clathrin adaptor AP-2 [59, 61]. Ingle et al. reported that CD19-directed mAb and mAb-drug conjugates are internalized by dynamin-dependent, clathrin-mediated endocytosis and their cellular uptake as well as intracellular trafficking mimic the endocytic pathway of transferrin [63]. Their studies demonstrated that anti-CD19 mAb are transported to late endosomes and lysosomes within 3 h [63]. Clathrin inhibitor chlorpromazine has been shown to inhibit the uptake of both transferrin and anti-CD19 mAb [64]. Detailed immunoelectron microscopy studies by Pulczynski et al. showed that upon binding of anti-CD19 mAb, CD19 antigen on the surface of pre-B ALL cells is internalized within 30 min via plasmalemmal pits, transferred through the endosomal compartment within 2 h, delivered to multivesicular bodies/late endosomes and lysosomes after 2h, and recycled [64]. Notably, the internalized anti-CD19 mAb-CD19 complexes showed close physical association with/attachment to the endosomal membrane [64]. This close association of internalized anti-CD19 mAb with the endosomal membrane likely facilitates the endosomal leakage and/or escape of chemical or biological substances attached to them into the cytosol and provides a cogent explanation for the documented potency of toxin conjugates [65], drug conjugates [66], as well as anti-sense oligonucleotide conjugates [67] of anti-CD19 mAb against CD19+ leukemia/lymphoma cells. Therefore, we propose that siRNA nanoparticles decorated with a CD19-specific biotargeting moiety would exhibit a significantly improved selective anti-cancer potency against CD19+ B-lineage leukemias and lymphomas.
References
- D.Haussecker.The Business of RNA Therapeutics in 2012. Molecular Therapy-Nucleic Acids, 2012, 2, e8; doi: 10.1038/mtna.2011.9.
- K.A. Whitehead, R. Langer, D.G. Anderson, Knocking down barriers: advances in siRNA delivery. Nature Reviews: Drug Discovery, 2009, 8:129-133
- JC. Burnett, JJ. Rossi, and K. Tiemann, Current progress of siRNA/shRNA therapeutics in clinical trials, Biotechnol. J. 6 (2011) 1130–1146
- Y-K, Oh and T. G. Park, siRNA delivery systems for cancer treatment, Advanced Drug Delivery Reviews, 61 (2009) 850–862
- D. Castanotto and JJ. Rossi, The promises and pitfalls of RNA-interference-based therapeutics, Nature. 22 (2009) 457
- J. Li, and L. Huang, Targeted delivery of RNAi therapeutics for cancer therapy Nanomedicine, 5:10 (2010) 1483–1486
- M. Dominska and DM. Dykxhoorn, Breaking down the barriers: siRNA delivery and endosome escape, Journal of Cell Science 123:8 (2010) 1183-1189.
- J. Wang, Z. Lu, MG. Wientjes, and JL-S. Au, Delivery of siRNA Therapeutics: Barriers and Carriers, The AAPS Journal, 12:4 (2010) 492-503
- S. Guo and L. Huang, Nanoparticles Escaping RES and Endosome: Challenges for siRNA Delivery for Cancer Therapy, Journal of Nanomaterials, Article ID 742895 (2011) 1-12
- W. Gao, et al., Progress in siRNA Delivery Using Multifunctional Nanoparticles, M. Sioud (ed.), RNA Therapeutics, Methods in Molecular Biology, 2010, 629, Springer Science+Business Media, Chapter 4: 53-67.
- M.E. Davis, J.E. Zuckermann, C.H.J. Choi, D. Seligson, A. Tolcher, C.A. Alabi, Y. Yen, J.D. Heidel, A. Ribas. Evidence of RNAi in humans from systemically administered siRNA via targeted nanoparticles. Nature, 2010, 464: 1067-1070
- EE. Salcher and E. Wagner, Chemically Programmed Polymers for Targeted DNA and siRNA Transfection, Top Curr Chem 296 (2010) 227–249
- ME. Davis and NC. Bellocq, Cyclodextrin-Containing Polymers for Gene Delivery, Journal of Inclusion Phenomena and Macrocyclic Chemistry, 44 (2002) 17–22
- DW. Pack, A.S. Hoffman, S. Pun, P.S. Stayton. Design and Development of Polymers for Gene Delivery, Nature Review, Drug Discovery, 2005, 4: 581-593
- P. Vader, L.J. van der Aa, G. Storm, R.M. Schiffelers, J.F. Engbersen, Polymeric carrier systems for siRNA delivery. Curr Top Med Chem. , 2012, 12:108-119.
- SD. Li and L. Huang, Targeted Delivery of Antisense Oligodeoxynucleotide and Small Interference RNA into Lung Cancer Cells, ACS, Molecular Pharmaceutics, 3:5 (2006) 579-588
- J. Li, Y. C. Chen, Y. C. Tseng, S. Mozumdar, and L. Huang, Biodegradable calcium phosphate nanoparticle with lipid coating for systemic siRNA delivery,” Journal of Controlled Release, 142:3 (2010) 416–421
- XW. He, T. Liu, Y.X. Chen, D.J. Cheng, X.R. Li, Y. Xiao, Y.L. Feng, Calcium carbonate nanoparticle delivering vascular endothelial growth factor-C siRNA effectively inhibits lymphangiogenesis and growth of gastric cancer in vivo, Cancer Gene Therapy, 2008, 15: 193–202
- X. Ming, Cellular delivery of siRNA and antisense oligonucleotides via recepto-mediated endocytosis, Expert Opin Drug Deliv, 2011, 8:435-49
- R.L. Juliano, X. Ming, O. Nakagawa, Cellular uptake and intracellular trafficking of antisense and siRNA oligonucleotides. Bioconjugate Chemistry, 2012, 23: 147-157
- http://clinicaltrials.gov/ct2/show/NCT01591356
- CN. Landen, A. Chavez-Reyes, R. Bucana C, Schmandt, M.T. Deavers, G. Lopez-Berestein, A.K. Sood. Therapeutic EphA2 gene targeting in vivo using neutral liposomal small interfering RNA delivery. Cancer Res., 2005, 65: 6910–6918
- WM. Merritt , Y.G. Lin , W.A. Spannuth , M.S. Fletcher , A.A. Kamat , L.Y. Han, C.N. Landen , N. Jennings , K. De Geest , R.R. Langley , G. Villares , A. Sanguino , S.K. Lutgendorf , G. Lopez-Berestein , M.M. Bar-Eli , A.K. Sood . Effect of Interleukin-8 Gene Silencing With Liposome-Encapsulated Small Interfering RNA on Ovarian Cancer Cell Growth, JNCI, 2008, 100:359-372
- M. Scherr, K. Battmer, T. Winkler, and O. Heidenreich, A. Ganser, M. Eder, Specific inhibition of bcr-abl gene expression by small interfering RNA. Blood, 2003, 101: 1566–1569
- M.A. Rahman , A.R. Amin , X. Wang , J.E. Zuckerman , C.H. Choi, B. Zhou, D. Wang , S. Nannapaneni , L. Koenig , Z. Chen , Z.G. Chen , Y. Yen , M.E. Davis , D.M. Shin , Systemic delivery of siRNA nanoparticles targeting RRM2 suppresses head and neck tumor growth. J Control Release. 2012, 159:384-92
- N. Manjunath, D.M. Dykxhoorn, Advances in Synthetic siRNA Delivery, Discovery Medicine, 2010, 9:418-430
- A. Eguchi, B.R. Meade, Y-C. Chang et al., Efficient siRNA delivery into primary cells by a peptide transduction domain-dsRNA binding domain fusion protein. Nature Biotechnology, 2009, 27:567-571
- E. Song, P. Zhu, S.K. Lee et al., Antibody mediated in vivo delivery of small interfering RNAs via cell surface receptors. Nature Biotechnology, 2005, 23:709-717
- J. Winkler, Nanomedicines based on recombinant fusion proteins for targeting siRNA oligonucleotides. Ther Deliv, 2011, 2: 891-905
- S.C. Semple, A. Akinc, J. Chen, A.P. Sandhu, B.L.L. Mui, C.K. Cho et al., Rational design of cationic lipids for siRNA delivery. Nature Biotechnology, 2010, 28: 172-176
- A. Akinc, et al., A combinatorial library of lipid-like materials for delivery of RNAi therapeutics, Nature Biotechnology, 26:5 (2008) 561-569
- KT. Love, et al., Lipid-like materials for low-dose in vivo gene silencing, PNAS, 107:5 (2010) 1864–1869
- A. Akinc, et al., Targeted Delivery of RNAi Therapeutics With Endogenous and Exogenous Ligand-Based Mechanisms, Molecular Therapy vol. 18:7 (2010) 1357–1364
- A. Akinc, M. Goldberg, J. Qin, J.R. Dorkin, C. Gamba-Vitalo, M. Maier et al., Development of lipidoid-siRNA formulations for systemic delivery to the liver. Molecular Therapy 2009, 17: 872-879
- S.C. Semple, A. Akinc, J. Chen, A.P. Sandhu, B.L.L. Mui, C.K. Cho et al., Rational design of cationic lipids for siRNA delivery. Nature Biotechnology, 2010, 28: 172-176
- K.A. Afonin, W.W. Grabow, F.M. Walker, E. Bindewald, M.A. Dobrovolskaia, B.A. Shapiro et al., Design and self-assembly of siRNA-functionalized RNA nanoparticles for use in automated nanomedicine. Nat Protoc. 2011, 6:2022-34.
- J. Lee, J. Hong, D.K. Bonner, Z. Poon, P.T. Hammond, Self-assembled RNA interference microsponges for efficient siRNA delivery. Nat Mater. 2012, 11:316-322.
- Y. Sakurai, H. Hatakeyama, Y. Sato, H. Akita, K. Takayama, S. Kobayashi et al., Endosomal escape and the knockdown efficiency of liposomal-siRNA by the fusogenic peptide shGALA. Biomaterials. , 2011, 32:5733-5742.
- S. Nimesh, Polyethylenimine as a Promising Vector for Targeted siRNA Delivery. Curr Clin Pharmacol. 2012 Mar 20. [Epub ahead of print]
- P. Liu , H. Yu , Y. Sun , M. Zhu , Y. Duan, A mPEG-PLGA-b-PLL copolymer carrier for adriamycin and siRNA delivery. Biomaterials. 2012 Mar 19. [Epub ahead of print]
- A. Alhaddad, M.P. Adam , J. Botsoa , G. Dantelle, S. Perruchas , T. Gacoin, C. Mansuy , S. Lavielle , C. Malvy , F. Treussart , J.R. Bertrand, Nanodiamond as a vector for siRNA delivery to Ewing sarcoma cells. Small. 2011, 4:7:3087-95.
- J. Sparks, G. Slobodkin, M. Matar, R. Congo, D. Ulkoski, A. Rea-Ramsey et al., Versatile cationic lipids for siRNA delivery. J Control Release. 2012, 158:269-76.
- S.W. Kim, N.Y. Kim, Y.B. Choi, S.H. Park, J.M. Yang, S. Shin, RNA interference in vitro and in vivo using an arginine peptide/siRNA complex system. J Control Release, 2010, 143: 335-343
- LB. Jeffs, et al., A scalable, extrusion-free method for efficient liposomal encapsulation of plasmid DNA, Pharmaceutical Research, 2005, 22:362-372
- Y. Gutierrez-Puente, et al., Safety, Pharmacokinetics, and Tissue Distribution of Liposomal P-Ethoxy Antisense Oligonucleotides Targeted to Bcl-21, J. Pharmaco. Exp. Therapeutics, 1999, 291:865-869
- S. Y. Wu, Development of a Novel Method for Formulating Stable siRNA-Loaded Lipid Particles for In vivo Use, Pharmaceutical Research, 2009, 26:512-522
- B. Stark, G. Pabst, R. Prassl. Long-term stability of sterically stabilized liposomes by freezing and freeze-drying: Effects of cryoprotectants on structure. Eur J Pharm Sci, 2010, 41: 546-55
- B. Y. Kim, J. T. Rutka, W. C. Chan. Nanomedicine. N Engl J Med, 2010, 363: 2434-2443.
- R. A. Petros, J. M. DeSimone, Strategies in the design of nanoparticles for therapeutic applications. Nat Rev Drug Discov, 2010, 9: 615-627
- K. Greish, Enhanced Permeability and Retention (EPR) Effect for Anticancer Nanomedicine Drug Targeting, Cancer Nanotechnology, Methods in Molecular Biology, 2010, 624: 25-37
- V. Torchilin, Tumor delivery of macromolecular drugs based on the EPR effect, Advanced Drug Delivery Reviews, 2011, 63: 131–135
- M.E. Davis, Z. Chen, D.M. Shin, Nanoparticle therapeutics: an emerging treatment modality for cancer. Nature Review Drug Discovery, 2008, 7:771-82
- K. Cho, X. Wang, S. Nie, Z.G. Chen, D.M. Shin. Therapeutic nanoparticles for drug delivery in cancer. Clin Cancer Res, 2008, 14:1310-6.
- D.E. Owens, N.A. Peppas, Opsonization, biodistribution, and pharmacokinetics of polymeric nanoparticles. Int J Pharm, 2006, 307:93-102
- Li WM, L. Mayer, M.B. Bally. Prevention of antibody-mediated elimination of ligand-targeted liposomes by using poly(ethylene glycol)-modified lipids. J Pharm Exp Ther, 2002, 300: 976-983
- J.E. Zuckerman, C.H. Choi, H. Han, M.E. Davis, Polycation-siRNA nanoparticles can disassemble at the kidney glomerular basement membrane, Proc Natl Acad Sci U S A., 2012, 109:3137-42
- SH. Yiv and FM. Uckun, Lipid spheres as attractive nanoscale drug delivery platforms for cancer therapy, J Nanomedic. Nanotechnol., 2012, 3:1-6
- R.K. Jain, T. Stylianopoulos, Delivering nanomedicine to solid tumors. Nat Rev Clin Oncol, 2010, 7: 653–664
- A.S. Narang, S. Varia, Role of tumor vascular architecture in drug delivery, Adv. Drug Delivery Reviews, 2011, 63: 640–658
- F.M. Uckun, W. Jaszcz, J. L. Ambrus, A.S. Fauci, K. Gajl-Peczalska, C.W. Song, M.R. Wick, D.E. Myers, K. Waddick, J.A. Ledbetter, Detailed studies on expression and function of CD19 surface determinant by using B43 monoclonal antibody and the clinical potential of anti-CD19 immunotoxins. Blood., 1988, 71: 13-29.
- F.M. Uckun, Regulation of human B-cell ontogeny (Review). Blood, 1990, 76: 1723-1733
- F.M. Uckun, A.L. Burkhardt, L. Jarvis, X. Jun, B. Stealey, I. Dibirdik, D.E. Myers, L. Tuel-Ahlgren, J.B. Bolen, Signal transduction through the CD19 receptor during discrete developmental stages of human B-cell ontogeny. J Biol Chem , 1993, 268: 21172-21184
- O.W. Press, A.G. Farr, K.I. Borroz, S.K. Anderson, P.J. Martin, Endocytosis and Degradation of Monoclonal Antibodies Targeting Human B-Cell Malignancies. Cancer Research, 1989, 49: 4906-4912
- G.S. Ingle, P. Chan, J.M. Elliott, W.S. Chang, H. Koeppen, J-P. Stephan, S.J. Scales, High CD21 expression inhibits internalization of anti-CD19 antibodies and cytotoxicity of an anti-CD19 drug conjugate. British Journal of Haematology, 2007, 140:46-58
- S. Pulczynski, A.M. Boesen, O.M. Jensen, Antibody-induced modulation and intracellular transport of CD10 and CD19 antigens in human B-cell lines: An immunofluorescence and immunoelectron microscopy study. Blood, 1993, 81: 1549-1557
- F.M. Uckun, K.J. Gajl-Peczalska, J.H. Kersey, L.L. Houston, D.A. Vallera, Use of a novel colony assay to evaluate the cytotoxicity of an immunotoxin containing pokeweed antiviral protein against blast progenitor cells freshly obtained from patients with common B-lineage acute lymphoblastic leukemia. J Exp Med. 1986, 163:347-68.
- F.M. Uckun, W.E. Evans, C.J. Forsyth , K.G. Waddick, L.T. Ahlgren, L.M. Chelstrom et al. Biotherapy of B-cell precursor leukemia by targeting genistein to CD19-associated tyrosine kinases. Science., 1995, 267:886-91.
- F.M. Uckun, S. Qazi, I. Dibirdik, D.E. Myers, Rational Design of an Immunoconjugate for Selective Knock-Down of Leukemia-Specific E2A-PBX1 Fusion Gene Expression in Human Pre-B Leukemia, Integrative Biology, 2012, in press





