A Rare Case of Haemophagocytic Syndrome in a HIV Seropositive Patient with Cytomegalovirus Induced Paraparesis
Chatterji S1*, Chakraborty S1, Shashya S Khaling1, Naskar A2, Ghosh MK2, Pal S2, Misra AK3, Mallik S4, Saha B5
1. Junior Resident, Department of Tropical Medicine, School of Tropical Medicine, Kolkata, India.
2. Assistant Professor, Department of Tropical Medicine, School of Tropical Medicine, Kolkata, India.
3. Clinical Tutor, Department of Tropical Medicine, School of Tropical Medicine, Kolkata, India.
4. Associate Professor, Department of Tropical Medicine, School of Tropical Medicine, Kolkata, India.
5. Professor & Head of Department of Tropical Medicine, School of Tropical Medicine, Kolkata, India.
*Corresponding Author
Dr. SoumyadipChatterji,
Junior Resident, Department of Tropical Medicine,
School of Tropical Medicine, Kolkata,
India Pin Code- 700073.
Tel: 919051851337
E-mail: dr.sdip@gmail.com
Received: July 09, 2015; Accepted: November 28, 2015; Published: December 03, 2015
Citation: Chatterji S, et al., (2015) A Rare Case of Haemophagocytic Syndrome in a HIV Seropositive Patient with Cytomegalovirus Induced paraparesis. Int J AIDS Res. 02(5), 39-42. doi: dx.doi.org/10.19070/2379-1586-150009
Copyright: Chatterji S© 2015. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
Abstract
Hemophagocytic syndrome or hemophagocytic lymphohistiocytosis (HLH ) is a rare but potentially life threatening condition characterized by uncontrolled activation of CD8+ T lymphocytes and macrophages that leads to organ damage. HLH can be primary (familial or genetic) and secondary. Secondary HLH occurs due to infections caused mostly by viruses like Ebstein Barr virus, Cytomegalovirus (CMV), Parvo B19 etc. Early diagnosis and treatment is the key to management of HLH.
We present here a case of a 30 year old female who presented with fever, jaundice, cough and progressive weakness of both lower limbs. There was no girdle like sensation, no bladder/bowel involvement. She was recently diagnosed to be seropositive for Human Immuno-deficiency Virus (HIV)-1 and had a CD4 count of 27/μL. She had hepatomegaly, pancytopenia, hyperbilirubinemia, hypoalbuminemia, raised liver enzymes and hypertriglyceridemia. CSF had raised protein, reduced sugar and neutrophils. DNA PCR for CMV was positive and there was evidence of CMV disease on fundoscopy. Her bone marrow revealed presence of haemophagocytes. She was treated successfully with Ganciclovir , Dexamethasone and anti-retroviral drugs and was discharged.
Though often fatal and difficult to diagnose, HLH is potentially treatable disorder.
2.Introduction
3.Case Report
5.Discussion
6.Conclusion
7.References
Keywords
Hemophagocytic Syndrome; HLH; HIV; CMV.
Introduction
Hemophagocytic syndrome or Hemophagocytic lymphohistiocytosis (HLH ) is a rare [1] but potentially life threatening condition characterized by uncontrolled activation of CD8+ T lymphocytes and macrophages that leads to organ damage [2]. Pediatric age group is most commonly affected in primary HLH but it can occur at any age [3, 4]. The manifestations of secondary HLH are often induced by an infection (which maybe viral, bacterial or parasitic), malignancy or autoimmune disease [4]. HLH can be familial (autosomal recessive) as well [3, 5]. The diagnosis is made by the identification of characteristic features of: Fever, hepatomegaly, cytopenias, hypertriglyceridemia or hypofibrogenemia, hemophagocytes in the marrow or other reticuloendothelial system organs, low or absent NK cell activity, a high ferritin level and a high soluble CD25 [6]. Although reactive haemophagocytic syndrome (RHS) has been reported in HIV-infected patients, we have little information about the features of this disorder in the era of combination antiretroviral therapy (ART) [7].
Case Report
A 30 years old female from East Midnapur, West Bengal, was admitted with high grade intermittent fever of 10 days, followed by jaundice which developed 2-3 days later. She had cough with expectoration of 1 week. She also had progressive weakness of both lower limbs for last 1 week, with no girdle like sensation over trunk. There was no bladder or bowel involvement or paraesthesia. Furthermore she also had recurrent genital ulcers (vesicular), for past 4 months. There was no past history of tuberculosis. There was no significant medical or surgical history preceding this, other than being diagnosed seropositive for Human Immunodeficiency Virus (HIV) a couple of months ago. Her CD4 cell
count was 27/μL. She was subsequently initiated on anti-retroviral therapy (ART) with Tenofovir, Lamivudine and Efavirenz (TDF+ 3TC+EFV) one month before the present illness.
On examination, she was conscious, alert, cooperative but was febrile, pale and icteric. Her vitals were stable (Pulse 110/min, BP 110/76 mm of Hg). She had hepatomegaly but there was no splenomegaly or ascites. She had crepitations on both lung bases. Power in her lower limbs was bilaterally 3/5. There was absence of knee and ankle jerk. Plantar was bilaterally flexor. Her initial reports revealed pancytopenia, hyperbilirubinemia, hypoalbuminemia, raised liver enzymes and hypertriglyceridemia. Renal function tests and electrolytes were normal. Peripheral blood smear did not reveal malaria parasite. Blood culture did not show any growth. Serology was negative for Leptospira, Dengue and Chikungunya. Serum toxoplasma IgG was negative too. Sputum did not reveal acid-fast bacillus (AFB), but showed growth of Pseudomonas aeruginosa in culture. Treatment of her lower respiratory tract infection was started with Piperacillin and Tazobactum along with Azithromycin [8] and the cause of her paraparesis was being searched for. Cerebrospinal fluid (CSF) study revealed decreased glucose, markedly elevated proteins with polymorphonuclear leukocytosis. CSF Cryptococcal antigen was negative, no AFB was seen nor was any fungus or malignant cell visible. CMV DNA was detected in real time-DNA PCR (polymerase chain reaction) from blood sample.
Ophthalmological examination was done, which revealed features of CMV retinitis. Her serum ferritin was very high (4238.6 ng/ml). Bone marrow aspiration and cytological examination revealed presence of haemophagocytes (Figure1). Multi-nucleate giant cells [9] could not be visualised as to be suspected in this case of HIV-CMV co-infection. Thus, the provisional diagnosis was Hemophagocytic syndrome due to CMV infection in a HIV patient. The neurological disease could be explained by CMV disease which caused the polyradiculopathy. With the presence of bilateral lower limb weakness in absence of any bladder/bowel involvement without upper level of sensory loss, we wanted to exclude the possibility, though unlikely, of HIV myelopathy - but the patient didn’t consent to MRI because of claustrophobia. She was also very sick. Diagnosis of peripheral neuropathy and ascending neuromuscular paralysis were excluded clinically.
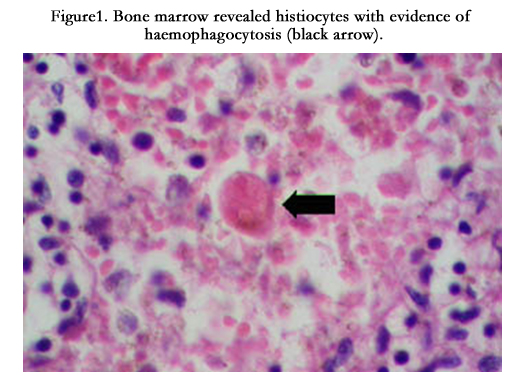
Figure 1. Bone marrow revealed histiocytes with evidence of haemophagocytosis (black arrow).
Patient was put on Injection Dexamethasone 100mg/m2 for 1st 3 days, which was tapered off over next 5 days as per HLH-94 protocol [10, 11]. Injection Ganciclovir 5mg/kg q12 hr was started for CMV disease which was the cause of HLH [12]. Injection G-CSF was given as well.
There was rapid improvement in the patient’s condition with subsidence of fever and improvement of all the laboratory parameters in a couple of weeks. Lower limb motor weakness improved slowly with physiotherapy and the patient was discharged after almost 1 month of her admission in an ambulatory state with a final diagnosis of paraparesis and HLH cause by CMV infection in a HIV patient. The prompt response to therapy proved the diagnosis.
Discussion
The hemophagocytic syndrome was first described in 1979 in immunosuppressed patients with viral infections [13]. It is a lifethreatening immune dysregulatory syndrome caused by severe hypercytokinemia due to a highly stimulated but ineffective immune process [11]. Despite recent gain in knowledge, the pathogenesis of HLH is unclear. However, it is known that the clinical manifestations of HLH are due to hyperactivation of CD8+ T lymphocytes and macrophages; proliferation, ectopic migration, and infiltration of these cells into various organs; hypercytokinemia with persistently elevated levels of multiple proinflammatory cytokines resulting in progressive organ dysfunction that may lead to death.
These interrelated factors underlie the clinical manifestations of prolonged fever, hepatosplenomegaly, bleeding, skin rash, CNS abnormalities, jaundice, and the laboratory findings of bicytopenia or pancytopenia, coagulopathy, hyperlipidemia, hypofibrinogenemia, hyperferritinemia, transaminitis, hyperbilirubinemia, and hypoalbuminemia [3]. HLH can be primary (familial or genetic) and secondary. Primary HLH usually present in childhood particularly < 2 years of age [14], though late presentations usually in 9 to 17 years have been reported. Secondary HLH occurs due to various causes including infections caused mostly by viruses like Ebstein Barr virus (EBV), Cytomegalovirus (CMV), Parvovirus B19 etc (Table 1)[15]. Whereas primary HLH is associated with perforin mutation [16], secondary HLH is associated with production of high levels of activating cytokines by host lymphocytes and monocytes. There is no test available that can differentiate between primary and secondary HLH. Course of the disease in both groups is not so varied but secondary HLH has good outcome if detected early [17].
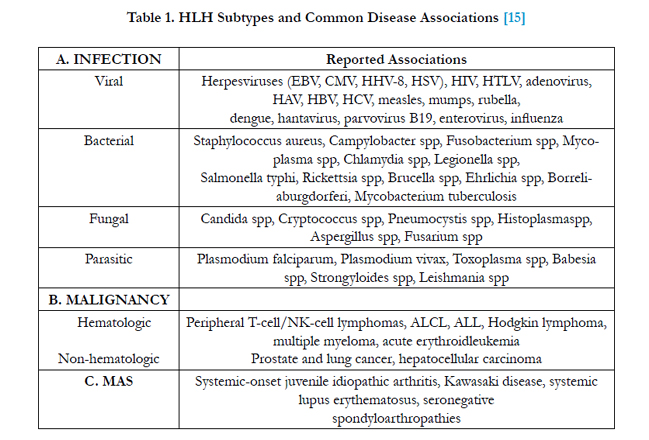

Table 1. HLH Subtypes and Common Disease Associations [15]
To diagnose HLH 5 out of 8 of the following criteria set up by Histiocyte Society 2004 (Table 2)[18] must be present. Our patient fulfils 5 criteria as she had fever, hypertriglyceridemia,pancy topenia, bonemarrow evidence of hemophagocytosis and hyperferritinemia.
EBV is the most common etiology, while CMV is associated with 30% to 40% of all virus-associated HLH cases [19]. Although the other factors could cause HLH, we believe that CMV was the main etiologic agent involved in this case. The prompt response to Ganciclovir and clinical and hematological improvement along with subsidence of paraparesis proved our suspicion. CMV is also known to causes transverse myelitis [20, 21] which is probably not the case here. This case emphasizes the importance of bone marrow examination and extensive investigation for opportunistic infections in immunocompromised patients presenting with febrile pancytopenia. In most cases, treatment of the underlying condition promotes complete remission of the clinical picture.

Table 2. Revised Diagnostic Guidelines for HLH [18]
Conclusion
This case report intends to alert treating physicians about the possible diagnosis of hemophagocytic syndrome whenever they are treating critically ill patients with variety of pertinent symptoms. Early diagnosis and treatment is the key to management of HLH. Underlying cause is to be detected in early course of illness to bring down the mortality and morbidity. Awareness regarding its clinical presentation and a high level of clinical suspicion are essential for diagnosis, especially inimmunocompromised patients. Moreover, active search for and identification of opportunistic infections at the time of diagnosis of HIV is essential.
References
- Aricò M, Janka G, Fischer A, Henter JI, Blanche S, et al. (1996) Hemophagocytic lymphohistiocytosis. Report of 122 children from the International Registry. FHL Study Group of the Histiocyte Society. Leukemia 10(2): 197-203.
- Filipovich AH (2009) Hemophagocytic lymphohistiocytosis (HLH) and related disorders. Hematology Am Soc Hematol Educ Program 2009: 127- 131.
- Weitzman S (2011) Approach to Hemophagocytic Syndromes. Hematology Am Soc Hematol Educ Program 2011: 178-183.
- Garett JP, Fung I, Rupon J, Knight A, Mizesko M, et al. (2012) Presentation of hemophagocytic lymphohistiocytosis due to a novel MUNC 13–4 mutation masked by partial therapeutic immunosuppression. Pediatr Rheumatol Online J 10: 13.
- Dufourcq-Lagelouse R, Jabado N, Le Deist F, Stéphan JL, Souillet G, et al. (1999) Linkage of familial hemophagocytic lymphohistiocytosis to 10q21- 22 and evidence for heterogeneity. Am J Hum Genet 64(1): 172-179.
- Bhattacharya M, Ghosh MK (2008) Hemophagoctic Lymphohistiocytosis – Recent Concept. J Assoc Physicians India 56: 453-457.
- Fardet L, Lambotte O, Meynard JL, Kamouh W, Galicier L, et al. (2010) Reactive haemophagocytic syndrome in 58 HIV-1-infected patients: Clinical features, underlying diseases and prognosis. AIDS 24(9): 1299-1306.
- Mendell LA, Wunderink RG, Anzueto A, Bartlett JG, Campbell GD, et al. (2007) Infectious Diseases Society of America/American Thoracic Society Consensus Guidelines on the Management of Community-Acquired Pneumonia in Adults. Clin Infect Dis 44(Suppl 2): S27-72.
- Bélec L, Gray F, Mikol J, Scaravilli F, Mhiri C, et al. (1990) Cytomegalovirus (CMV) encephalomyeloradiculitis and human immunodeficiency virus (HIV) encephalitis: presence of HIV and CMV co-infected multinucleated giant cells. Acta Neuropathol 81(1): 99-104.
- Kleynberg RL, Schiller GJ (2012) Secondary Hemophagocytic Lymphohistiocytosis in Adults: An Update on Diagnosis and Therapy. Clin Adv Hematol Oncol 10(11): 726-732.
- Jordan MB, Allen CE, Weitzman S, Filipovich AH, McClain KL (2011) How I treat hemophagocytic lymphohistiocytosis. Blood 118(15): 4041- 4052.
- Guidelines for the prevention treatment of opportunistic infections in HIVinfected adults and adolescents. (2013) http://aidsinfo.nih.gov/guidelines
- Risdall RJ, McKenna RW, Nesbit ME, Krivit W, Balfour HH Jr, et al. (1979) Virus-associated hemophagocytic syndrome: a benign histiocytic proliferation distinct from malignant histiocytosis. Cancer 44(3): 993-1002.
- Ericson KG, Fadeel B, Nilsson-Ardnor S, Söderhäll C, Samuelsson A, et al. (2001) Spectrum of perforin gene mutations in familial hemophagocytic lymphohistiocytosis. Am J Hum Genet 68(3): 590-597.
- Rosado FG, Kim AS (2013) Hemophagocytic Lymphohistiocytosis: An Update on Diagnosis and Pathogenesis. Am J Clin Pathol 139(6): 713-727.
- Stepp SE, Dufourcq-Lagelouse R, Le Deist F, Bhawan S, Certain S, et al. (1999) Perforin gene defects in familial hemophagocytic lymphohistiocytosis. Science 286(5446): 1957-1959.
- Fisman DN (2000) Hemophagocytic syndromes and infection. Emerg Infect Dis 6(6): 601-608.
- Henter JI, Horne A, Aric´o M, Egeler RM, Filipovich AH, et al. (2007) HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer 48(2): 124-131.
- Créput C, Galicier L, Buyse S, Azoulay E (2008) Understanding organ dysfunction in hemophagocytic lymphohistiocytosis. Intensive Care Med 34(7): 1177-1187.
- Karacostas D, Christodoulou C, Drevelengas A, Paschalidou M, Ioannides P, et al. (2002) Cytomegalovirus-associated transverse myelitis in a non-immunocompromised patient. Spinal Cord 40(3): 145-149.
- Rafailidis PI, Mourtzoukou EG, Varbobitis LC, Falagas ME (2008) Severe cytomegalovirus infection in apparently immunocompetent patients: a systematic review. Virol J 5:47.





