Mitochondrial D Loop Variants in Juvenile Myoclonic Epilepsy Patients: A Study from Asian Indians
Maniyar Roshan Z1,2*, Doshi MA3, Jahan P4
1 Research Scholar, Department of Anatomy, Krishna Institute of Medical Sciences, Karad, Maharashtra, India.
2 Sandor Lifesciences Pvt. Ltd., Banjara Hills, Hyderabad, Telangana, India.
3 Professor and Head, Department of Anatomy, Krishna Institute of Medical Sciences, Karad, Maharashtra, India.
4 Associate Professor, Department of Zoology, Moulana Azad National Urdu University, Hyderabad, Telangana, India.
*Corresponding Author
Roshan Zameer Maniyar,
Research Scholar, Department of Anatomy,
Krishna Institute of Medical Sciences, Karad, Maharashtra, India
and Sandor Lifesciences Pvt. Ltd., Banjara Hills, Hyderabad, Telangana, India
Tel: 9241730949
E-mail: roshan.anatomist@gmail.com
Received: August 31, 2017; Accepted: November 27, 2017; Published: November 29, 2017
Citation: Maniyar Roshan Z, Doshi MA, Jahan P. Mitochondrial D Loop Variants in Juvenile Myoclonic Epilepsy Patients: A Study from Asian Indians. Int J Genomics Proteomics Metabolomics Bioinformatics. 2(3), 36-42.
Copyright: Maniyar Roshan Z© 2017. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
Abstract
Background: Mitochondrial mutations may exert unfavorable effect on neuron synapses which may lead to the loss of motor functions causing seizures. Mitochondrial displacement loop (D-loop) is the hotspot for mtDNA alterations which influence the generation of cellular reactive oxygen species (ROS). Lack of cellular energy (ATP) due to defective oxidative phosphorylation (OXPHOS) and ROS can cause somatic mutations in mtDNA. The concern of the present study is to understand the mitochondrial basis of the disease in our population by identifying the Novel mitochondrial mutations which in turn may facilitate the diagnosis of a section of Juvenile myoclonic epilepsy (JME) patient’s thereby better management of the condition.
Most neurodegenerative diseases is abnormal folding and these changes in normal protein metabolism often lead to neuronal cell death and failure of the affected regions of the central nervous system. Abnormal protein accumulation may be attributed to dysfunctional mitochondria and damage by reactive oxygen species. No treatments have yet been found successful to rid a person of neurodegeneration completely.
Methods: The two hypervariable segments (HV1 and HV2) of mtDNA D-Loop region were sequenced in 35 unrelated JME patient’s blood samples. The mtDNA samples were amplified by polymerase Chain Reaction (PCR), and then SNPs were detected by direct sequencing and comparing the sequencing results with the mtDNA Cambridge Reference Sequence.
Result: We detected multiple mutations in D-loop region including Pyrimidine transitions 27%, 56%; Purine transitions 23%, 39% and Transversion 50%, 5% in HV1 and HV2 segments respectively. Overall we observed 64 mutations in different nucleotide positions in HV1 and HV2 segments of D-loop region and 18 mutations in HV2 segments in the JME patients.
Conclusion: The study deal with the sequencing of HV1 and HV2 regions of mtDNA D- loop region in JME patients of Asian Indians. We found several novel mutations, suggesting these mutations may be involved in the pathogenesis of JME as many neurological disorders are associated with mitochondrial mutations and mitochondrial dysfunction. Further studies including normal healthy individuals from same ethnicity may help in identifying the role of these genetic variations in the pathogenesis of JME.
2.Introduction
3.Prevalence
4.Material and Method
4.1 Study Design
4.2 Ethical Approval
5.Methods of Technique
5.1 Sample Collection
5.2 DNA Extraction
5.3 Isolation of Genomic DNA
5.4 Genotype by PCR
5.5 Agarose Gel Electrophoresis (AGE)
5.6 Statistical Analysis
5.7 mtDNA D-loop Polymorphism
6.Results
6.1 Mutation Analysis of HV1 and HV2 Segments
6.2 Polymorphism in Hypervariable Segment-1 (HVS1)
6.3 Polymorphism in Hypervariable Segment-2 (HVS2)
7.Discussion
8.Conclusion
9.Acknowledgements
10.References
KeyWords
mtDNA; Hypervariable Region, D-Loop; Juvenile Myoclonic Epilepsy; ROS.
Introduction
Epilepsy is a frequent manifestation of mitochondrial disorders, as the central nervous system (CNS) is often severely affected in these diseases. Seizures are commonly seen in Alpers-Huttenlocher syndrome, Leigh syndrome, MELAS syndrome (mitochondrial encephalo myopathy, lactic acidosis and stroke-like episodes), MEMSA syndrome (myoclonic epilepsy, myopathy, sensory ataxia) and in ataxia neuropathy spectrum. Pathogenic mtDNA mutations are often heteroplasmic so that both the mutant variant and the wild type variant are present in the mitochondria. The proportion of the mutant variant must exceed a certain threshold before symptoms of mitochondrial disease become manifest. Furthermore, mitochondrial oxidative stress may play a role in epileptogenesis by virtue of affecting neuronal excitability.
The human mitochondrial DNA or genome (mtDNA) is a circular 16.6 kb double-stranded molecule, has been completely sequenced and all the mitochondrial encoded genes have been identified and characterized. The human mtDNA is a 16569 bp, which encodes 13 proteins (from 37 genes) involved in respiratory chain, 2rRNAs, and a set of 22 tRNAs that are essential for protein synthesis in the mitochondria [1, 2]. The outside strand of double helical circular DNA is called as heavy strand (H-strand) and the inside one is light strand (L-strand). The H-strand and the L-strand contain 28 and 9 genes, respectively. Virtually, all normal cells consist of high copy numbers of mitochondria (103- 104 copies per cell) and the vast majority of copies are identical at birth.
Mitochondrial oxidative phosphorylation is the principle source of ATP in neurons and adequate levels of ATP are essential to maintain the neuronal plasma membrane potential via the sodium–potassium ATPase, which consumes about 40% of the energy. ROS are produced as by-products of respiration and the oxidative phosphorylation process. Mitochondrial abnormalities have been recognized as important contributors to an array of neuromuscular diseases including Juvenile myoclonic epilepsy (JME) [3, 8]. Maniyar Roshan Z et al., (2017) analyzed 75 JME patients ranging in age from 7 to 41 years (mean 20 years) revealed that seizures usually occur shortly after awakening and precipitated by sleep deprivation [4]. Damaged mtDNA molecules can result in malfunctioning proteins and altered mtDNA replication and/ or transcription efficiency. The D-loop region is a hot spot for mtDNA changes, and posses’ non-coding region which consist of two hyper variable regions i.e HVR1 at nucleotides 16024- 16383 and HVR2 at nucleotides 42-498 positions [5, 6]. High mutation rates of mtDNA are caused by lack of protective histones, inefficient DNA repair systems and continuous exposure to mutagenic effects of oxygen radicals generated by oxidative phosphorylation [7]. MtDNA exhibit high mutation rate, haplogroup, no recombination and is maternally inherited through eggs cytoplasm.
Two nonsense mutations may leads to protein is incomplete and observed in BRD2 gene. These types of mutations change a triplet codon that encodes a stop codon. The nuclear DNA (nDNA) sequence shortened the protein products of BRD2 gene due to stop codons [9].
Huoponen et al., (2001) studied regarding the mitochondrial DNA variation in Australian population. Direct sequencing of the control region (HVSI) of the mtDNA revealed 34 distinct sequences. The hypervariable regions I, II and III of the Malaysian population showed that these regions of mtDNA could be used in forensic investigations complement anthropology and population studies in Asia.
The analysis of the genetic structure of the Kazakh population as estimated from mitochondrial DNA polymorphism showed that Western Europe (55%) and Eastern Europe (41%) mtDNA lineages exist in the Kazakhs population and high genetic diversity was observed in the Kazakhs population. However, there are no studies in relation to JME and mtDNA mutations though mitochondrial function is associated with neurological disorders such as epilepsy. The purpose of present study is to evaluate the mutation frequencies in the two hypervariable segments (HV1 and HV2) of the mtDNA D-loop region using PCR and sequencing techniques in JME patients from south India.
Prevalence
The prevalence rate of pathogenic mtDNA mutations was detected to be more than 1 in 200 live births [10]. The minimum prevalence rate for mtDNA mutations was 1 in 5,000 (20 per 100,000). The exact prevalence of mtDNA diseases is notoriously difficult to ascertain due to clinical heterogeneity of mitochondrial diseases and causative mutations. Data on a single-point mutation in Finland population indicated that approximately 1 in 6,000 individuals are affected. British population estimates that about 1 in 3,500 people either have mtDNA disease or are at risk of developing it [11, 12].
The present study was carried out from 2003-2015 in the Department of Anatomy, Krishna Institute of Medical Sciences University, Karad (M.H) and research work conducted in collaboration with Bioserve Biotechnologies (India) private Ltd. Mallapur, Hyderabad (T.S) and Sandor proteomics Private Ltd, Banjara Hills, Hyderabad (T.S).
All the participants were evaluated under the supervision of Neurologist who was specialized in epilepsy.
A human studies protocol was approved by the Research Ethical committee, Krishna Institute of Medical Science Deemed University, Karad, M.H, and India.
All aspects of the study were explained, and written informed consent was obtained before the study was initiated and in case of minor participants consent from parents or guardians was obtained.
We drawn 5 ml of blood from 35 (20 males and 15 females) samples were obtained from Telangana, Andhra Pradesh and Karnataka (Hyderabad-Karnataka region) of three states of South India. The blood samples collected based on the diagnostic evidence confirmation by the Neurologist. The blood was transferred into EDTA tubes and sends to Sandor proteomics Private Ltd, Banjara Hills, Hyderabad (T.S) and stored at 4°C till further use and core mtDNA sequence in D-loop region completed at Bioserve Biotechnologies (India) private Ltd. Mallapur, Hyderabad.
Genomic DNA was extracted from peripheral blood leukocytes by using the standard phenol-chloroform method. All single nucleotide polymorphisms (SNPs) were analyzed by PCR followed by Sanger sequencing. The PCRs were performed with 40 ng of genomic DNA; the PCR products were checked on 2% agarose gels and were directly sequenced using Big Dye Terminator Cycle Sequencing Ready Reaction Kit and the ABI PRISM 3730 DNA analyzer.
The present study included control amplification in each step to assess any possible contamination. Negative control amplification, to check the purity of the PCR reagents with no DNA added. Blank control amplification is used to check the purity of the extraction reagents. Positive control amplification used to monitor the success of thermocycler reactions. Forward and reverse primers were designed and conditions optimized to amplify and sequence the template mtDNA by Big Dye terminator cycle sequencing. The results were compared with the Cambridge reference sequence. The sequence and position of the forward (F) and reverse (R) primers for PCR and primer pairs were used for amplification of HVR1 and HVR2 sequencing are as follows:
PCR was performed in a total volume of 25 (mu) L, and the hotstart technique was applied to the reactions of both segments. The PCR was performed in a 50 μl reaction volume mix containing 40 ng of template DNA, 6.25 pmol of each primer, 200 lM of dNTPs, 1.5 mM MgCl2, 1• reaction buffer and 0.3 U of Taq pol enzyme (Bangalore Genei, India). The following conditions were used for 30 cycles: denaturation at 94°C for 1 min, followed by annealing at 56°C for 1 min for HVR-1 and 58°C for 1 min for HVR-II, and extension at 72°C for 1 min, repeated for 30 cycles followed by a final extension step at 72°C for 5 min.
Agarose gel electrophoresis was used to examine the quality and quantity of extracted DNA. The amplified PCR products were checked by electrophoresis using 2% agarose gel with ultraviolet illumination run at 110V for 15 minutes after ethidium bromide staining. The PCR product of gel plate placed in alpha imager through UV rays and the image was visualized on monitor for analysis of the quality of the primers.
Participant characteristics, mitochondrial D-loop variants and copy number were described as mean ± SD by Microsoft statistical software for windows. Chi-square tests were used to compare frequency of mitochondrial D-loop variants in JME patients, and D-loop variants numbers. P<0.05 was considered statistically significant.
The mtDNA HVR1 and HVR2 sequences of each individual were compared with the revisited Cambridge Reference Sequence (rCSR) by using the Applied Biosystems Seq Scape ver. 2.5 Software. Sequence analysis was performed using an ABI PRISM 377 genetic analyzer sequences instrument. Mutations were confirmed by repeated analyses from both H and L strands.
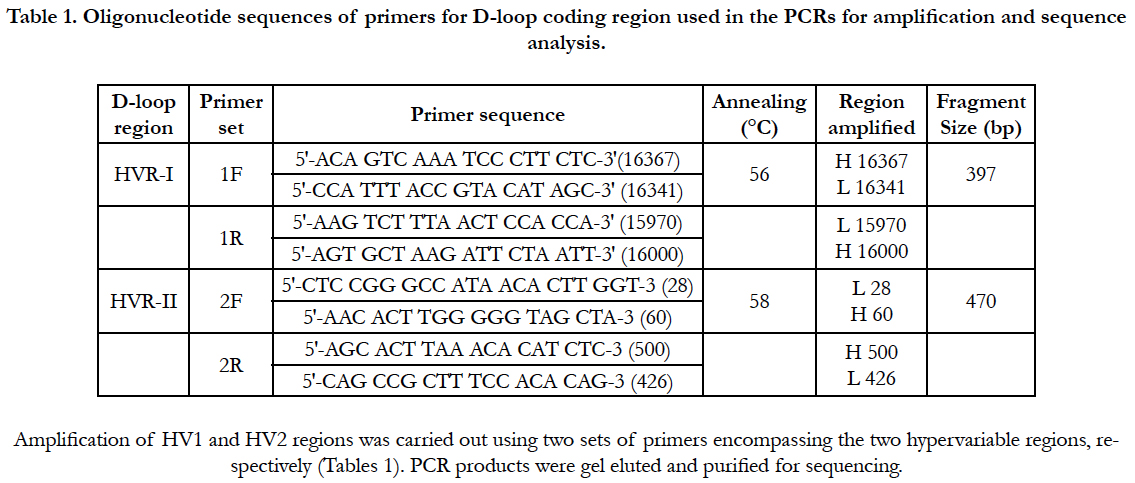

Results
To best of our knowledge, this is the first report to examine the involvement of mitochondrial mutations in Juvenile Myoclonic Epilepsy. PCR products from successful amplifications were subjected to mtDNA sequencing in D-loop region of HV1 and HV2 segments (Figure 1). The sequence electro-pherogram of mtDNA showed polymorphisms in different nucleotide position in HVR-1 and HVR-2 segments (Table:2). The samples were not showing any heteroplasmy which confirms that our mtDNA is not contaminated.
Mitochondrial D-loop mutations from HV-1 and HV-2 segments observed 121 in different genotypes in 25 unrelated juvenile myoclonic epilepsy donors (Table: 2).
Hydroxyl radicals also delaminate nucleotides, leading to Point mutations notably base pair changes C>T, G>C and G>T observed. The base substitution C>T and T>C are termed as a transition, because C and T are both pyrimidine nucleotides (Table: 2). The A>C, C>A, A>T, C>G, G>C and G>T changes are termed as transversions, because a purine nucleotide (G) is converted into a pyrimidine (C or T).
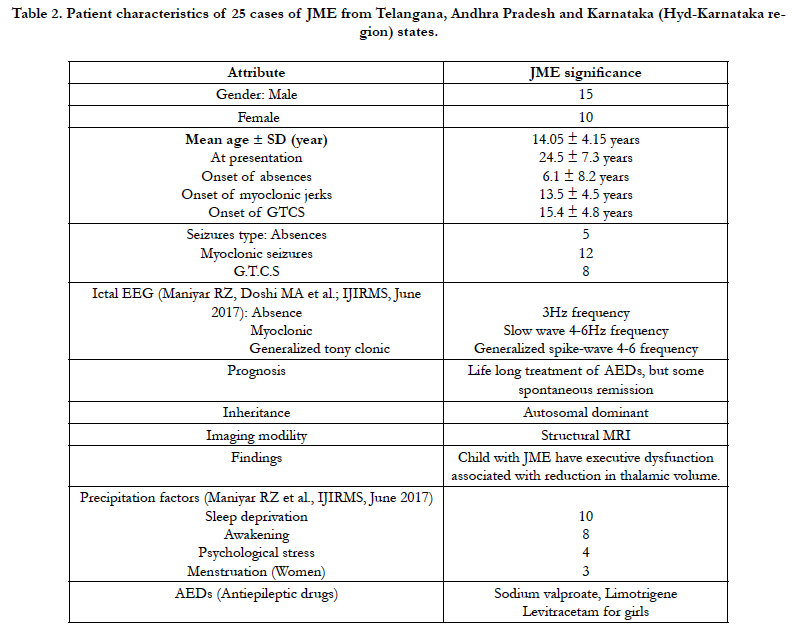
In the present study, we carried out mtDNA sequencing in two of the hypervariable segments (HVR1 and HVR2) of mitochondrial genome among 35 JME patients. Alignments were made with the Cambridge Reference Sequence (CRS) for human mitochondrial DNA (mtDNA)
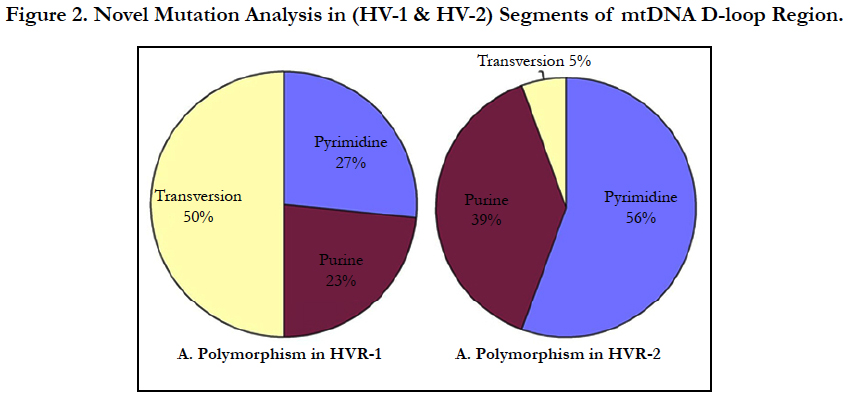
Mutations, often multiple, including pyrimidine transitions (27%), purine transitions (23%) and Transversion (50%) in HV1 segment, while HV2 segment consist pyrimidine transitions (56%), purine transitions (39%) and one polymorpmism of Transversion (5%) (Figure 3). Large number of variations observed particularly transversion between HV1 and HV2 segments (Figure 3).
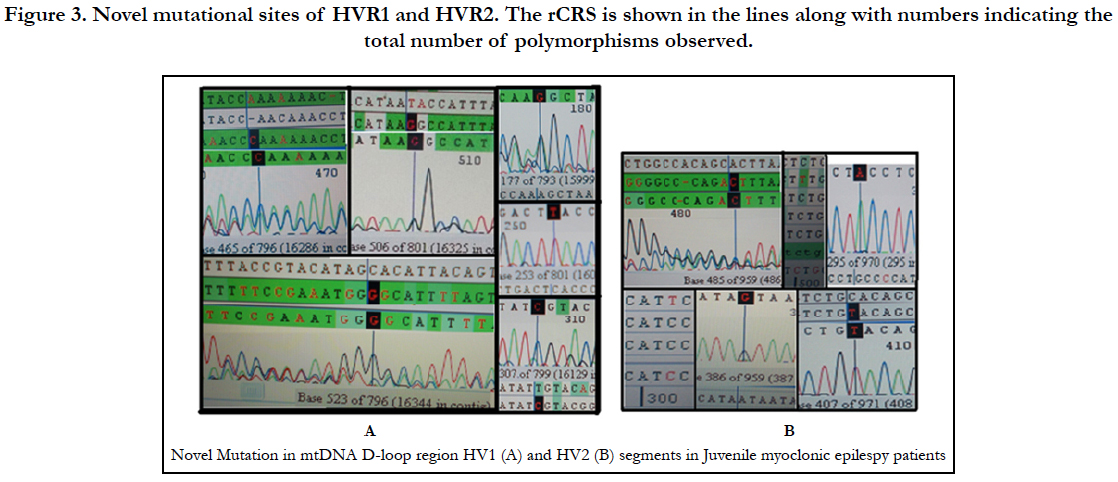

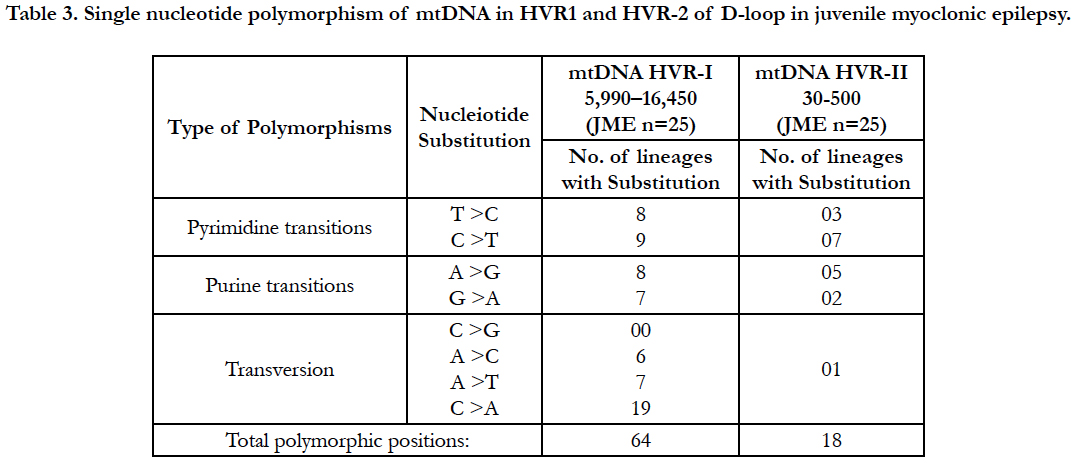
Analysis of mitochondrial nucleotide positions (5,990-16450) in HVRI and HVR II (nucleotide positions: 30-500) showed the presence of a large number of nucleotide base variations. The HVR1 sequences among the 35 JME donors revealed a total of 64 polymorphic sites found in HV1 segment. Some of the novel mutations are not reported previously. The majority of the novel mutations observed in are Transversions (32/25) C>A, A>T, and A> C (Figure 4). The nucleiotide substitution A>T (transversion) was previously published.
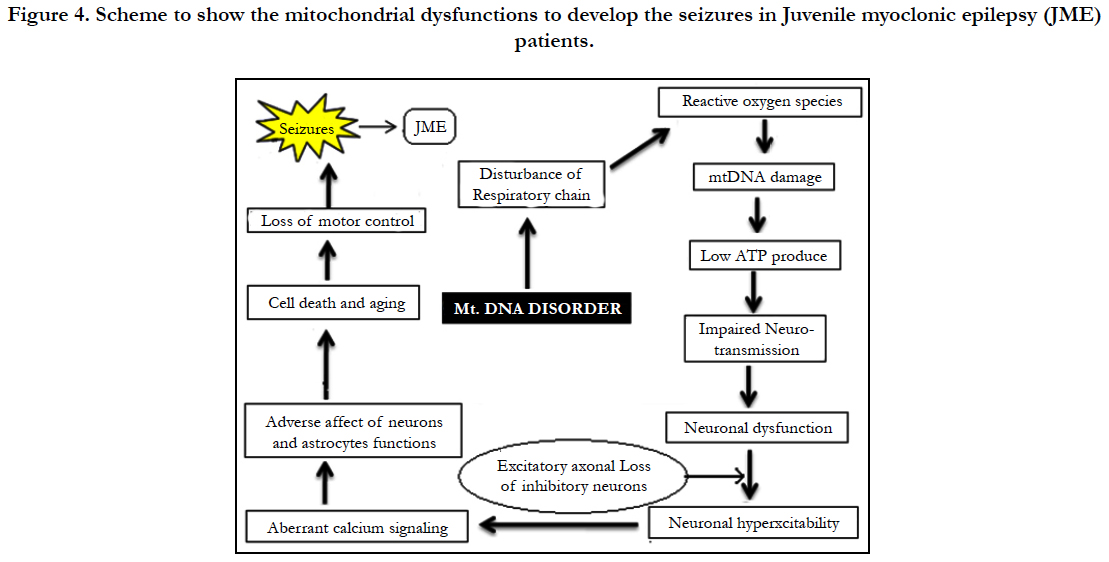
The pyrimidine transition changes observed are T>C, and C>T (Table: 2). Soodyall et al., 1996, and Piercy et al., 1993, identified A>G purine transition (Figure 4). These novel mutations could perhaps provide important insights into the impact of mtDNA changes during seizures.
Polymorphic analysis of mtDNA D-loop region in HV2 segment revealed 18 polymorphic sites found in JME cases (Figure 4) including pyrimidine, purine and transversion mutations. The segment of HV2 sequences revealed 8 new polymorphic sites are not reported previously. The sequence polymorphism of pyrimidine transitions changes detected are C→T, and T→C and purine transitions changes A→G, and G→A (Figure 4). We detected only one transversion novel mutation A>T identified in this region (Table 2). Vigilant et al., 1991; Piercy et al., 1993; Bertranpetit et al., 1995; Sajantila et al., 1995 reported that the observed sequences differed from those of the Cambridge Reference Sequence (CRS) at a number of positions.
Discussion
As of now there are no reports to estimate the frequency of polymorphisms found in mitochondrial DNA associated with juvenile myoclonic epilepsy. To the best of our knowledge, this is the first report to examine the involvement of mitochondrial mutations in JME patients. A search of literature failed to turn up any relevant genetic based studies. The study of mtDNA is important for medical specialties and biologists because it does not undergo recombination and exhibits matrilineal inheritance. The hyper variability (HV) define high mutation rate (7±10 times faster) compared with genomic (nuclear) DNA. Researchers from the Gladstone Institutes have showed impaired mitochondria can lower cellular energy levels and lead to neuronal dysfunction. D-loop is a non-coding region of mitochondrial DNA which constitutes about 7% of the mitochondrial genome and associated with regulatory sequences.
Investigations dealing with KCN mouse model support a the link between ROS formation due to increased brain lipid peroxidation and the pathogenesis of seizures [13]. MtDNA mutations were found to accumulate in high levels in dividing cells and presumably stem cells 95%. mtDNA mutations of all inherited human D-loop polymorphisms are transitions [14]. Our data showed a high frequency polymorphism of mitochondrial D-Loop mutations in HV1 segment. Based on our results it is presumed that mtDNA mutations within the two hypervariable (HV1 and HV2) D-Loop control regions may contribute to the events in JME patients. Zsurka and Kunz from Bonn University, Germany, recently published in the journal Lancet Neurology that there is a relationship between mitochondrial dysfunction and epilepsy. Suggesting, mitochondrial dysfunction as an important contributor to neuromuscular and neurodegenerative diseases. In the brain, the mitochondria are responsible for providing ATP for neurotransmission, reactive oxygen species (ROS) signaling at synapses, and regulating presynaptic and postsynaptic calcium concentrations [19]. Mitochondria help the cells to maintain proper concentration of calcium ions and play important role in the process of apoptosis or programmed cell death. The mitochondrial genome codes for 13 proteins, all essential for the normal function of mitochondria. Abnormal protein accumulation may be attributed to dysfunctional mitochondria and damage by reactive oxygen species. ROS could change the shape of proteins completely, which would an effect on protein functions and neurons in central nervous system (CNS). A high concentration of reactive oxygen species in mitochondria can induce DNA damage . The brain is essential for high energy demands and mitochondria play a central role in calcium homeostasis by sequestering calcium, which is an important signaling molecule participating in several cellular pathways under physiological conditions [15]. The mitochondria in a cell are keeping calcium levels stable, and dysfunction of the mitochondria may cause spikes in calcium levels. The excess of calcium can lead to cell death. When calcium levels fluctuate in the cell, apoptosis is usually initiated because more calcium creates increased sensitivity to “apoptotic stimuli”. However that impaired calcium handling resulting from mitochondrial dysfunction can increase neuronal excitability, leading to seizures [16, 17]. Sequence polymorphism of mtDNA in HVR I and HVR II can be used as a genetic marker for genetic analysis. Aberrant calcium signaling in astrocytes and neurons is likely to be a factor in epileptogenesis, probably by causing excessive synchronization of neurons [18]. Roshan Z et al., (2017) analyzed 75 JME patients ranging in age from 7 to 41 years (mean 20 years) revealed that seizures usually occur shortly after awakening and precipitated by sleep deprivation [4]. Epileptogenesis is characterized by neuronal hyper excitability that may be triggered by multiple molecular and physiological changes [20]. Indeed, in a recent study in human cells, a strong correlation was found between mitochondrial respiration and mitochondrial mutagenesis. Seizures could be due to changes in calcium levels that might affect neuronal excitability, synaptic transmission and neurotransmitters. The mtDNA is known for “large scale deletions or point mutations” that cause adverse effects to the mitochondria function by negatively affecting energy production processes. Penta et al., (2000) reported that alterations in expression of mitochondrial DNA (mtDNA) encoded polypeptides required for oxidative phosphorylation and cellular ATP generation may be a general characteristic of cancer cells. Howell et al., (2000) analyzed the transmission of the human mitochondrial genome and the transmission of heteroplasmic mtDNA sequence changes from one generation to the next often involves rapid shifts in allele frequency.
One attractive hypothesis is that damage to the mitochondrial genes could increase production of reactive oxygen species, not only leading to further mutations within the mtDNA but also increasing the instability of nuclear DNA or mtDNA mutation [21].
Conclusion
To the best of our knowledge this is the first study dealing with the sequencing of HV1 and HV2 regions of mt DNA D loop region in JME patients of Asian Indians. We found several novel mutations, suggesting these mutations may be involved in the pathogenesis of JME as many neurological disorders are associated with mitochondrial mutations and mitochondrial dysfunction.
The limitations of our study are firstly, we do not have the data of normal individuals of our population to compare with that of JME patients, hence, difficult to differentiate between the disease causing mutations from that of the common polymorphisms present in our population. However, we observed high mutations frequency as well as novel mutations that are not reported previously when compared with Cambridge Reference Sequence. In view of these facts, further studies including normal healthy individuals may help in identifying the role of these genetic variations in the pathogenesis of JME. In addition using bioinformatics tools to investigate the interaction of the variant sites with the replication machinery and the regulatory molecules will help in speculating the influence of these mutations in JME.
Acknowledgements
We thank all of the JME patients and case subjects for participating in this study. We sincere thanks to Dr. (Mrs) Gonwker, KIMSU, Karad (M.H); and Dr. (Mrs) Doshi M.A; Professor and Head, KIMS, Karad (M.H); we thankful to Dr. (Mrs). Parveen Jahan, Associate Professor, Dept. of Zoology, Moulana Azad National Urdu University, Hyderabad, (T.S) for the valuable discussion and guidelines.
I am grateful to Managing Director, Sandor Lifesciences, Pvt. Ltd. Banjara Hills, Hyderabad for giving permission to complete the thesis work.
We thank to Dr. B.N. Umarji Professor and Dr. Channanna.C Director, BRIMS, Bidar (K.A). I am very grateful to Dr. G. Shivannarayan, M.D, D.M (Neurologist), Dr. Syed Muneer, M.D (pathologist) for their great role to publish this article.
References
- DiMauro S, Schon EA. Mitochondrial DNA mutations in human disease. Am J Med Genet. 2001;106(1):18–26. PubMed PMID: 11579421.
- Thorburn DR, Dahl HH. Mitochondrial disorders: genetics, counseling, prenatal diagnosis and reproductive options. Am J Med Genet. 2001;106(1):102–114. PubMed PMID: 11579429.
- Andrews RM, Kubacka I, Chinnery PF, Lightowlers RN, Turnbull DN, Howell N. Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat Genet. 1999 Oct; 23( 2):147. PubMed PMID: 10508508.
- Maniyar RZ, Doshi MA, Parveen J, Umarji BN, Shivannarayan G, Parthasaradhi G, et al., Influence of Sleep Deprivation in Series of Seizures Pattern in JME Patients. IJIRMS. 2017 Jun 6;2(6):857-861.
- Suzuki M, Toyooka S, Miyajima K, Iizasa T, Fujisawa T, Bekele NB, et al. Alterations in the mitochondrial displacement loop in lung cancers. Clin Cancer Res. 2003 Nov 15;9(15):5636-5641. PubMed PMID: 14654546.
- Seyedhassani SM, Houshmand M, Kalantar SM, Modabber G, Aflatoonian A. No mitochondrial DNA deletions but more D-loop point mutations in repeated pregnancy loss. J Assist Reprod Genet. 2010 Nov;27(11):641–648. PubMed Central PMCID: PMC2995430.
- Miyazono F, Schneider PM, Metzger R, Warnecke-Eberz U, Baldus SE, Dienes HP, et al. Mutations in the mitochondrial DNA D-Loop region occur frequently in adenocarcinoma in Barrett's esophagus. Oncogene. 2002 May 23;21(23):3780–3783 PubMed PMID: 12032845. doi: 10.1038/ sj.onc.1205532.
- Tuppen HA, Blakely EL, Turnbull DM, Taylor RW. Mitochondrial DNA mutation and human disease. Biochim Biophys Acta. 2010 Feb;1797(2):113– 128. PubMed PMID: 19761752.
- Maniyar Roshan Z, Dosi MA, Proaveen J, Umarji BN, Shivanarayan G, Syed M, et al. Point Nonsense Mutations in BRD2 gene of Juvenile Myoclonic Epilepsy (JME) patients: A study from Dravidian Linguistic population in South Indian Population. 2017 Jun; 7(6).
- Elliott HR, Samuels DC, Eden JA, Relton CL, Chinnery PF. Pathogenic mitochondrial DNA mutations are common in the general population. Am J Hum enet. 2008 Aug ;83(2):254–260. PubMed PMID: 18674747.
- Majamaa K, Moilanen JS, Uimonen S, Remes AM, Salmela PI, Karppa M, et al. Epidemiology of A3243G, the mutation for mitochondrial encephalomyopathy, lactic acidosis, and strokelike episodes: prevalence of the mutation in an adult population. Am J Hum Genet. 1998 Aug; 63(2):447–454. PubMed Central PMCID: PMC1377301.
- Schaefer AM, Taylor RW, Turnbull DM, Chinnery PF. The epidemiology of mitochondrial disorders-past, present and future. Biochim Biophys Acta. 2004 Dec;659(2-3):115-120. PubMed PMID: 1557602.
- Yamamoto H, Tang HW. Preventive effect of melatonin against cyanideinduced seizures and lipid peroxidation in mice. Neurosci Lett. 1996 Mar 29; 207(2):89–92. PubMed PMID: 8731428.
- Lutz S, Weisser HJ, Heizmann J, Pollak S. Location and frequency of polymorphic positions in the mtDNA control region of individuals from Germany. Int J Legal Med. 1998;111(2): 67-77. PubMed PMID: 9541853.
- Llorente-Folch I, Rueda CB, Pardo B, Szabadkai G, Duchen MR, Satrustegui J. The regulation of neuronal mitochondrial metabolism by calcium. J Physiol. 2015 Aug 15;593(16): 3447-362. PubMed PMID: 25809592.
- Kunz WS. The role of mitochondria in epileptogenesis. Curr Opin Neurol. 2002 Apr;15(2):179-84.
- Kann O, Kovacs R. Mitochondria and neuronal activity. Am J Physiol Cell Physiol 2007 Feb;292(2):C641–57. PubMed PMID: 17092996.
- Steinlein OK. Calcium signaling and epilepsy. Cell Tissue Res. 2014 Aug;357(2):385–93. PubMed PMID: 24723228.
- Simeone KA, Matthews SA, Samson KK, Simeone TA. Targeting deficiencies in mitochondrial respiratory complex I and functional uncoupling exerts anti-seizure effects in a genetic model of temporal lobe epilepsy and in a model of acute Temporal lobe seizures. Exp Neurol. 2014 Jan;251:84–90. PubMed PMID: 24270080.
- Rowley S, Liang LP, Fulton R, Shimizu T, Day B, Patel M. Mitochondrial respiration deficits driven by reactive oxygen species in experimental temporal lobe epilepsy. Neurobiol Dis. 2015 Mar;75:151–8. PubMed PMID: 25600213.
- Bandy B, Davison AJ. Mitochondrial mutations may increase oxidative stress: Implications for carcinogenesis and aging? Free Radical Biol Med.1990;8(6):523-539. PubMed PMID: 2193852.





