Description of Chaperone Protein or Q9PEQ0 (HTPG_XYLFA)in Xylella Fastidiosa Organism by- BLAST, Jpred4, and FTMap Methods with help of Pymol Software
Reza Pashaei
Department of Biotechnology, chemistry and pharmacy, University of Siena, Italy.
*Corresponding Author
Reza Pashaei,
Department of Biotechnology, Chemistry and Pharmacy at University of Siena, Italy.
E-mail: reza.pashaei@student.unisi.it
Received: June 15, 2016; Accepted: July 14, 2016; Published: July 19, 2016
Citation: Reza Pashaei (2016) Description of Chaperone Protein or Q9PEQ0 (HTPG_XYLFA) in Xylella Fastidiosa Organism byBLAST, Jpred4, and FTMap methods with help of Pymol Software. Int J Genomics Proteomics Metabolomics Bioinformatics. 1(3), 14-16.
Copyright: Reza Pashaei© 2016. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
Abstract
Xylella fastidiosa is a fastidious, xylem-limited bacterium that causes a range of economically important plant diseases and Chaperone proteinor Q9PEQ0 is Xylella Fastidiosa organism that this article intends to know this protein three dimensional image. This research defines the information about Xylella Fastidiosa bacterium and homology image for UniProtKB - Q9PEQ0 (HTPG_XYLFA) or Chaperone protein HtpG with help of biology sites such as BLAST and Jpred4 which are relevant to PDB file of UniProt site. On the other hand, Pymol software utilized for showing the 3D image of this proteinnamely Chaperone protein with choosing the Zinc-83296662 for obtaining the Vina result after that, getting the FTMap result. The aim of this article is showing the three dimensional image of Chaperone protein in Pymol software, illustrates the result of processes that help to describe the Chaperone protein structure. Besides, compare the vina Pymol result and FTMap result. This article explain BLAST, Jpred4, and FTMap methods with help of Pymol software.
2.Introduction
3.General Protein Description
4.Materials and Methods
4.1.Homology Modeling and Methodology
4.2.Blast
4.3.Jpred4
4.4.Vina Procedure
4.5.FTMap result
5.Result
6.Conclusion
7.References
KeyWords
Xylella Fastidiosa; Protein Structure; Homology; Pymol.
Introduction
Xylella fastidiosa is a fastidious, xylem-limited bacterium that causes a range of economically important plant diseases [1]. This bacterium is related to Xanthomonadaceae family of X. fastidiosa Species. One of the most important effects of this bacterium is Phoney Peach disease but Xylella Fastidiosa has other bad impacts such as on the olive trees namely Olive Quick Decline Syndrome and Chlorosis disease in citrus. I determined about special protein namely Q9PEQ0 (HTPG_XYLFA) Protein in terms of structure, homology, Vina result, and FTMap that is relevant to Xylella Fastidiosa bacteria.
General Protein Description
UniProtKB - Q9PEQ0 (HTPG_XYLFA) or Chaperone protein HtpG is a protein of htpG gene that show in the Xylella fastidiosa (strain 9a5c) organism. The Alternative names of Chaperone protein are:
1) Heat shock protein HtpG
2) High temperature protein G
Chaperone protein has the long taxonomic lineage namely cellular organisms → Bacteria → Proteobacteria → Gammaproteobacteria → Xanthomonadales → Xanthomonadaceae → Xylella → Xylella fastidiosa. This protein has particular place in subcellular location, in fact, this place is Cytoplasm.
Materials and Methods
Homology modeling or comparative modeling of protein shows the experimental three dimensional (3D) structures of protein and amino acid sequence.
The BLAST programs include a feature for filtering the query sequence through programs that search for low-complexity regions [2]. For obtaining the Homology model for UniProtKB - Q9PEQ0 (HTPG_XYLFA) or Chaperone protein HtpG first of all, we utilized of UniProt namely Universal Protein Resource for finding the information about this protein because has complete data about proteins after that, with the use of BLAST namely Basic Local Alignment Search Tool that is a suite of programs provided by NCBI for aligning query sequences against present in a selected target database that results are reported in a form of a ranked list followed by a series of individual sequence alignments, plus various statistics and scores. The BLAST algorithm was developed as a way to perform DNA and protein sequence similarity searches by an algorithm that is faster than FASTA but considered to be equally as sensitive [2]. For illustrating the experimental evidence Q9PEQ0 (HTPG_XYLFA) protein level, I use of BLAST. First of all, I utilized UniProt site for downloading the BLAST file (www.uniprot.org/uniprot/Q9PEQ0) after the Job Status part, I obtained the result of BLAST namely finding the regions of local similarity between sequences as well as identify the members of gens families (blast.ncbi.nlm. nih.gov/Blast.cgi)
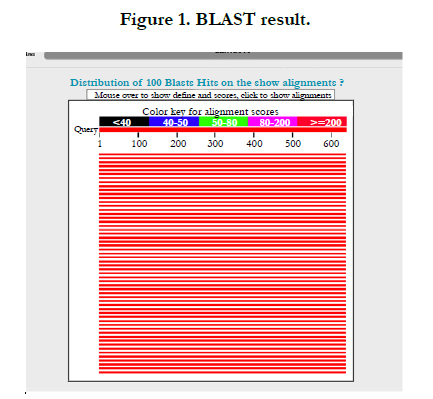

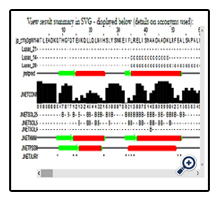
JPred4 is the latest version of the popular JPred protein secondary structure prediction server which provides predictions by the JNet algorithm, one of the most accurate methods for secondary structure prediction [3]. Accordingly, in order to maintain support for legacy courses and scripts, the results options in JPred4 include all the original formats and styles (PDF, HTML, etc.) as well as the intermediary processing files [3]. For predicting the second structure of Q9PEQ0 (HTPG_XYLFA) protein, I used the Jpred4 server that result of this server are SVG graph and full multiple sequence alignments
(www.compbio.dundee.ac.uk/jpred4/results/jp_z11g3gW/jp_z11g3gW.results.html).

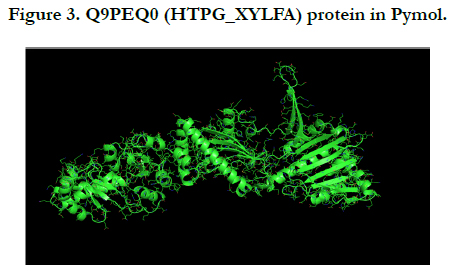
The above pictures illustrate the UniProtKB - Q9PEQ0 (HTPG_ XYLFA) or Chaperone protein HtpG in Pymol software by saving PNG image. The first image is primary image of protein and the second image is related to add the cartoon part for obtaining the clear image.
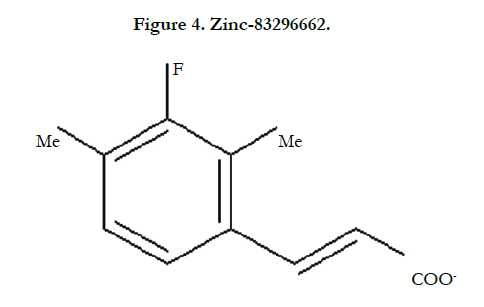
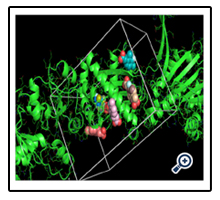
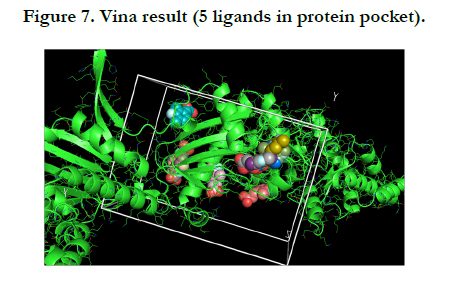
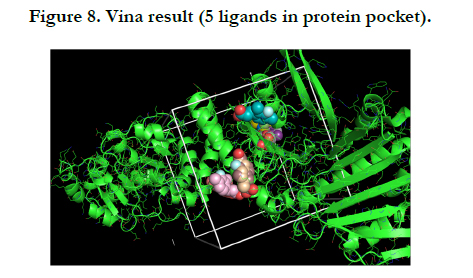
After choosing the molecule of Zinc docking site, and making the protein PDB file in the Pymol the Vina procedure started. My protein is UniProtKB - Q9PEQ0 (HTPG_XYLFA) or Chaperone protein HtpG and my molecule is Zinc-83296662 that popular name is (E)-3-(3-fluoro-2,4-dimethyl-phenyl) prop-2-enoic.
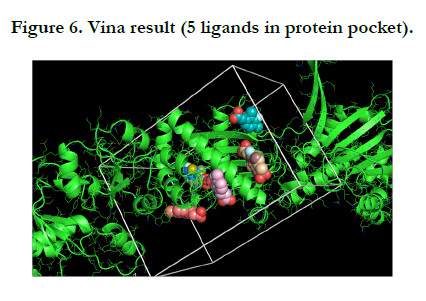
For this protein and molecule Vina had 5 results, summarized in the one PNG image for understanding very well.
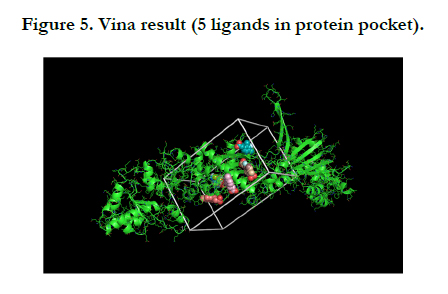

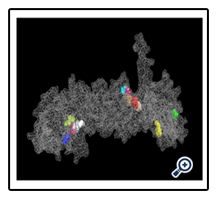
After showing the Vina results, we got the FTMap result (computational solvent mapping) namely recognizes the binding hot spots of macromolecule. The result defined that both of the user input and processed input was same and have favorable positions which showed the Chaperone protein structure.


FTMAP, a direct computational analog of the experimental screening approaches, globally samples the surface of a target protein using small organic molecules as probes, finds favorable positions, clusters the conformations and ranks the clusters on the basis of the average energy [4]. To use FTMap, users submit a protein, DNA or RNA structure in PDB (Protein Data Bank) format [5]. The images shown below describe the FTMap results with non-bonded interactions and Hbond interactions graphs.

Result
In this report, results of the Vina Pymol have five flexible ligands in protein pocket which are relevant to Zinc. On the other hand, FTMap Pymol has eleven results namely eleven hotspots that is difference between the Vina Pymol result and FTMap Pymol result.
Conclusion
In this report there are cycles of approaches for creating homology models and build a protein structure for the UniProtKB - Q9PEQ0 (HTPG_XYLFA) or Chaperone protein HtpG protein. In this case, BLAST can compare the primary biological sequence and secondary structure prediction namely defining α-Helices and β-sheet by Jpred4 and PHYRE2. On the other hand, HHpred can detect the protein homology and defines structure prediction but the created model is linear. Finally, the quality of protein structure model in this protein estimated by Qmean (Swiss Model) therefore, for this protein in the result Omean score is 0.6 and in the estimated absolute quality part Z-score is -1.81. Besides, Vina Pymol for Chaperone protein has result namely 5 aspects of ligandsbut FTMap in Pymol has 11 results.
References
- Simpson AJ, Reinach FC, Arruda P, Abreu FA, Acencio M, et al., (2000). The genome sequence of the plant pathogen Xylella fastidiosa. Nature 406(6792): 151-159. Doi:10.1038/35018003
- David W Mount (2007) Using the Basic Local Alignment Search Tool (BLAST). CSH Protoc pdb.top17. doi: 10.1101/pdb.top17.
- Alexey Drozdetskiy, Christian Cole, James Procter, Geoffrey J. Barton (2015) JPred4: a protein secondary structure prediction server. Nucleic Acids Research 43.
- Chi Ho Ngan, Tanggis Bohnuud, Scott E. Mottarella, Dmitri Beglov, Elizabeth A. Villar, et al., (2012) FTMAP: extended protein mapping with userselected probe molecules. Nucleic Acids Res 40: W271-w275. doi: 10.1093/nar/gks441
- Dima Kozakov, Laurie E Grove, David R Hall, Tanggis Bohnuud, Scott E Mottarella, et al., (2015) The FTMap family of web servers for determining and characterizing ligand-binding hot spots of proteins. Nat Protoc 10(5):733-755. doi: 10.1038/nprot.2015.043





