Primary Adrenal Insufficiency (PAI): A Major Teaching Hospital Experience, Riyadh, Saudi Arabia
Al-Jurayyan NA*
Professor and Pediatric Endocrinologist, Endocrine Division, Department of Pediatrics, College of Medicine and King Khalid University Hospital (KKUH), King Saud University Medical City, Riyadh, Saudi Arabia.
*Corresponding Author
Nasir A.M. Al Jurayyan,
Professor and Pediatric Endocrinologist,
Department of Pediatrics (39) College of Medicine and KKUH,
King Saud University, P.O. Box 2925,
Riyadh 11461, Saudi Arabia.
Tel: 966-11-467-1504
Fax: 966-11-467-1506
E-mail: njurayyan@gmail.com
Received: July 02, 2015; Accepted: August 13, 2015; Published: August 15, 2015
Citation: Al-Jurayyan NA (2015) Primary Adrenal Insufficiency (PAI): A Major Teaching Hospital Experience, Riyadh, Saudi Arabia. Int J Clin Ther Diagn 3(4), 92-94. doi: dx.doi.org/10.19070/2332-2926-1500018
Copyright: Al-Jurayyan NA© 2015. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
Abstract
Background: Primary adrenal insufficiency (PAI) in children is an uncommon, but potentially fatal. The current symptoms include weakness, fatigue, anorexia, abdominal pain, weight loss, orthostatic hypotension, salt craving and characterized by hyperpigmentation.
Material and Methods: This is a retrospective, hospital based-study, conducted at King Khalid University Hospital (KKUH), during the period January 1989 and December 2014. Review of medical record of patient diagnosed with primary adrenal insufficiency. The diagnosis was based on medical history, physical examination and low levels of glucocorticoids and raised adrenocorticotropic hormone (ACTH). Appropriate laboratory and radiological investigations were also reviewed.
Results: During the period under review, January 1989 and December 2014, a total of 125 patients with the diagnosis of primary adrenal insufficiency were seen. Inherited disorders like congenital adrenal hyperplasia and hypoplasia were common, 85.5%. However, variable autoimmune mediated etiologic diagnosis accounted for, 13%, were also seen. The appropriate various laboratory and radiological investigations should be planned.
Conclusion: Although, congenital adrenal hyperplasia was the commonest etiology, however, congenital adrenal hypoplasia should not be over looked. The diagnosis of PAI can be challenging in some patients, and therefore appropriate serological and radiological investigations should be done.
2.Introduction
3.Materials and Methods
4.Results
5.Discussion
6.Conclusion
7.References
Keywords
Etiology; Primary Adrenal Insufficiency; Saudi Arabia.
Introduction
Primary adrenal insufficiency (PAI) in children is an uncommon, but potentially fatal and life-threatening condition. The clinical symptoms of adrenal insufficiency include weakness, fatigue, anorexia, abdominal pain, weight loss, orthostatic hypotension, salt craving and characterized by hyperpigmentation. It comprises a heterogeneous group of both congenital and acquired disorders [1-6]. Congenital adrenal hyperplasia, is a group of autosomal recessive disorders resulting from the deficiency of one of the enzymes required to synthesis cortisol, was the commonest [7, 8]. Auto-immune induced adrenal insufficiency either isolated of as a part of autoimmune polyendocrine syndromes have been described, but the frequency of these occurrence in children with PAI has not been determined. Various mutations in genes, and autoantibodies to gland-specific target antigens such as 21-hydroxylase have been well described [9, 10].
The objective of this study was therefore, to define the etiology of PAI in a large referral, teaching hospital in Riyadh, Saudi Arabia.
Materials and Methods
All patients followed by the author at the Endocrine Unit at the King Khalid University Hospital between January 1989 and December 2014, with the diagnosis of primary adrenal insufficiency (PAI) were reviewed. The diagnostic categories retained were as follows: congenital adrenal hyperplasia (CAH), congenital adrenal hypoplasia, Addison’s disease, autoimmune polyendocrine syndromes, triple A syndrome (Allgrove syndrome), adrenoleukodystrophy and bilateral adrenal haemorrhage. Data reviewed included patient’s age, sex, clinical presentation, laboratory and radiological investigations. Congenital adrenal hyperplasia (CAH) was diagnosed as recommended [7].
The presence of anti-adrenal antibodies in serum was evaluated by indirect immunofluorescence. Mutational analysis of the AIRE gene was sequenced as described [9].
Results
During the period under review January 1989 and December 2014, a total of 125 patients were seen at the Endocrine Unit, King Khalid University Hospital, Riyadh, Saudi Arabia, with the diagnosis of primary adrenal insufficiency Table 1. There were 103 patients, 46 males and 57 females, with CAH aged between newborns to 13 years of age. Of these, 84 (81.1%) patients, 36 male and 48 females, were having CAH due to 21α-hydroxylase deficiency, with salt-wasting in 77 (91.7%) patients, 15 (14.6%) patients, 7 males and 8 females, patients with 11-β-hydroxylase deficiency which only 4 (3.9%) patients were due to 3-β-hydroxysteroid hydrogen deficiency, all were salt-waster. Table 2.
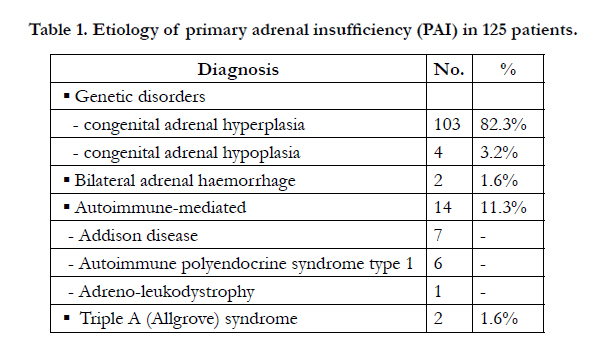

Table 1. Etiology of primary adrenal insufficiency (PAI) in 125 patients.

Table 2. Distribution of 103 patients with congenital adrenal hyperplasia (CAH) and enzyme deficiency.
Congenital adrenal hypoplasia was also diagnosed in 4 patients, from 2 families being presented with adrenal insufficiency with salt-wasting. Both families had history of neonatal deaths. Adrenal haemorrhage is relatively an uncommon condition with a variable and non-specific presentation seen in two patients. Figure 1, showing abdominal computed tomography (CT) revealing adrenal haemorrhage.
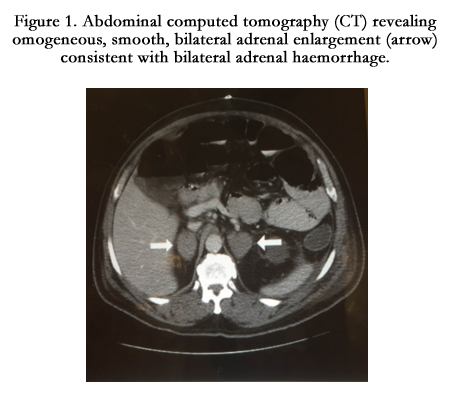
Figure 1. Abdominal computed tomography (CT) revealing homogeneous, smooth, bilateral adrenal enlargement (arrow) consistent with bilateral adrenal haemorrhage.
Adrenal insufficiency related to autoimmune disorders was the second most common, accounting to approximately 11%. Addison’s disease, as an isolated adrenal insufficiency in 7 patients, while in 6 patients it was associated with autoimmune polyendocrine syndrome Type 1 (APS-1).
X-linked adreno-leukodystrophy (ALD), an inherited disorder, due to the deficiency of ligoro-ceroyl-coA ligase (synthetase) enzyme, required for synthesis of long-chain-fatty acid was present in one patient. Figure 2 of magnetic resonance imaging (MRI) with characteristic changes. Allgrove syndrome, a rare autosomal disorder which is characterized by symptoms of alacrima, achalasia, and adrenal insufficiency.
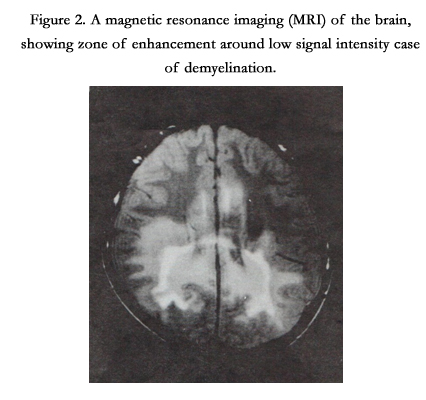
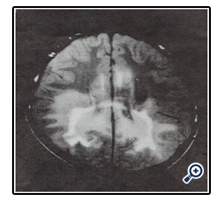
Figure 2. A magnetic resonance imaging (MRI) of the brain, showing zone of enhancement around low signal intensity case of demyelination.
Discussion
Adrenal insufficiency is the clinical manifestation of deficient production or action of glucocorticoids, with or without deficiency also in mineralocorticoids and adrenal androgen. It is a life-threatening disorder that can result from variable aetiologies, of which, primary adrenal failure was the commonest [1-6]. Congenital adrenal hyperplasia (CAH), is the common (82.3%) disorder, encountered in our study. It is an autosomal recessive disorder caused by reduced or complete absence of the enzymatic activities of steroid biosynthesis pathway [7]. In Saudi Arabia, this is not an unusual finding in a country with increased rate of consanguinity [11-13]. It should be differentiated from congenital adrenal hypoplasia. In contrast, congenital adrenal hypoplasia, is relatively a rare X-linked recessive disorder characterized by glucocorticoid and mineralocorticoid deficiency with low levels of androgen and normal external genitalia. Some rare cases develop adrenal failure only in adulthood [14, 15]. Bilateral adrenal haemorrhage is one of other causes of adrenal insufficiency presented in two infants, in this series. It is rare potentially life-threatening condition. The diagnosis is often complicated by its non-specific presentation and its tendency to intervene in stressful critical illness. Due to many coagulation disorders, haemorrhage is a major cause of morbidity and mortality. It can be diagnosed by ultrasonography, computed tomography (CT) and magnetic resonance imaging (MRI). The exact incidence of the condition is unknown. It is classically associated with meningococcemia (Waterhouse - Friderichsen syndrome), but may occur with any septic process. Traumatic events, burns, antiphospholipid syndrome, heparin associated thrombocytopenia, thrombophilic syndrome, anti-coagulated therapy, and abdominal surgery are some of the other causes of adrenal bleeding. The pathophysiology is still unclear. However, some particular anatomic features of the glands, such as a rich arterial blood supply that feeds into a dense and delicate sub-capsular capillary network [16, 17].
Autoimmune adrenal failure was the second leading cause of PAI, in our series, accounting for 11% of cases. It can present as an isolated condition or can be associated with other autoimmune disorders (autoimmune polyendocrine syndrome). Patients with autoimmune polyendocrine syndrome usually developed primary adrenal insufficiency earlier and had a history of chronic candidiasis and/or hypoparathyroidism. All patients with adrenal failure due to autoimmune syndrome had positive auto-adrenal antibodies [9, 18]. Adrenal failure was the common presentation in Xlinked adrenoleukodystrophy (ALD) [19, 20].
Conclusion
The diagnosis of primary adrenal insufficiency (PAI) can be challenging. Congenital adrenal hyperplasia was the commonest 82.3% diagnosis, with autoimmune disorders being the second found in 11%.
Acknowledgement
The author would like to thank Ms. Loida M. Sese for typing the manuscript and extend his thanks and appreciation to Miss Hadeel N. Al Jurayyan for her support and help in preparing this manuscript.
References
- Charmandari E, Nicolaides NC, Chrousos GP (2014) Adrenal insufficiency. Lancet 383(9935): 2152-2167.
- Nicolaides NC, Chrousos G, Charmandari E (2000) Adrenal insufficiency. MDText.com. Inc., South Dartmouth (MA).
- Oelkers W (1996) Adrenal insufficiency. N Engl J Med 335(16): 1206-1212.
- Arlt W, Allolio B (2003) Adrenal insufficiency. Lancet 361(9372): 1881-1893.
- Neary N, Nieman L (2010) Adrenal insufficiency, etiology, diagnosis and treatment. Curr Opin Endocrinol Diabetes Obes 17(3): 217-223.
- Husebye E, Lovas K (2009) Pathogenesis of primary adrenal insufficiency. Best Pract Res Clin Endocrinol Metab 23(2): 147-157.
- Al-Jurayyan NA (2015) Congenital adrenal hyperplasia in Saudi Arabia: The biochemical characteristics. Int J Health Sci Res 5(5): 98-102.
- Joint LWPES/ESPE` CAH Working Group (2001) Consensus statement on 21-hydroxylase deficiency from the Lawson Wilkins Pediatric Endocrine Society and the European Society for Pediatric endocrinology. J Clin and Endocrinol Metab 87(9): 4048-4053.
- Micheles AW, Gottlieb PA (2010) Autoimmune polyglandular syndromes. Nat Rev Endocrinol 6(5): 270-277.
- Meloni A, Perniola R, Faa V, Corvaglia E, Cao A, et al. (2002) Delineation of the molecular defects in the AIRE gene in autoimmune polyendocrinopathy- candidiasis-ectodermal dystrophy patients from Southern Italy. J Clin Endocrinol Metab 87(2): 841-846.
- Saedi-Wong S, Al-Frayh AR, Wong HY (1989) Socio-economic, epidemiology of consanguineous matings in the Saudi Arabian population. J AsianAfrican Stud 24(3-4): 247-251.
- El Mouzan MI, Al Salloum AA, Al Herbish AS, Qurachi MM, Al Omar AA (2008) Consanguinity and major genetic disorders in Saudi children: a community-based cross sectional study. Ann Saudi Med 28(3): 169-173.
- Al Jurayyan NA, Osman H (2015) The increased prevalence of congenital adrenal hyperplasia in Saudi Arabia: the role of consanguinity and multiple siblings involvement. Eur J Res Med Sci 3(1): 31-34.
- Al Jurayyan NA, Abdullah MA, Al Herbish AS, Abo Bakr AM (1995) Congenital adrenal hypoplasia: a disorder frequently missed. Ann Saudi Med 15(6): 563-565.
- Laverty CR, Fortune DW, Beischer NA (1973) Congenital idiopathic adrenal hypoplasia. Obstet Gynecol 41(5): 655-664.
- Rao RH (1995) Bilateral massive adrenal haemorrhage. Med Clin North Am 79(1): 107-129.
- Napier C, Pearce SH (2012) Autoimmune Addison’s disease. Press Med 41(12): e626-635.
- Nerup J (1974) Addison’s disease – clinical studies. A report of 108 cases. Acta Endocrinol 76(1): 127-141.
- Kemp S, Berger J, Aubourg P (2012) X-linked adrenoleukodystrophy: clinical, metabolic, genetic and pathophysiological aspects. Biochim Biophys Acta 1822(9): 1465-1474.
- Al Herbish AS, Salih MAM, Al Hussain M, Al Jurayyan NAM, Patel PJ, et al. (1993) X-linked adrenoleukodystrophy in the Arab ethnic group: presentation and management of a child. Med Sci Res 21: 439-441.





