Drug-eluting Stents in Premature Coronary Disease in Young People with Homozygous Familial Hypercholesterolemia and Prior Liver Transplant. A Report of Two Cases
Echeverri D*, Cabrales JR, Cardenas I, Ordonez F
Department of Hemodynamics and Cardiovascular Intervention, Fundación CardioInfantil – Instituto de Cardiología, School of Medicine – Universidad del Rosario, Bogotá, Colombia.
*Corresponding Author
Dr. Darío Echeverri M.D., F.A.C.C.,
Service Hemodynamics and Cardiovascular Interventions,
Cardioinfantil Foundation, Institute of Cardiology,
Street 163 A number 13B - 60. First Floor, Bogotá, Colombia.
Tel: (+57-1) 6791192/ (+57-1) 6672727, Extn: 1114
Fax: (+57-1) 6690382
E-mail: decheverri@cardioinfantil.org
Article Type: Case Report.
Recieved: May 11, 2015; Accepted: May 22, 2015; Published: May 28, 2015
Citation: Echeverri D, Cabrales JR, Cardenas I, Ordonez F (2015). Drug-eluting Stents in Premature Coronary Disease in Young People with Homozygous Familial Hypercholesterolemia and Prior Liver Transplant. A Report of Two Cases. Int J Cardiol Res. 02(1), 15-20. doi: dx.doi.org/10.19070/2470-4563-150004
Copyright: Echeverri D© 2015. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
2.Abbreviations
3.Introduction
4.Patients and Methods
5.Case 1
6.Case 2
7.Discussion
8.Conclusions and Recommendations
9.References
KeyWords
Atherosclerosis; Hypercholesterolemia; Stents; Liver; Transplantation.
Abbreviations
FH: Familialhypercholesterolemia; CD: Coronary disease; LDL-c: Low density lipoproteins
Introduction
Lipoprotein metabolism disorders, as well as high fat diets, obesity and physical inactivity have given rise to an atherosclerotic disease epidemic worldwide. The interaction of these adverse environmental factors with genetic and acquired lipoprotein disorders predispose to an increasingly earlier development of atherosclerosis. In the United States, mortality due to coronary artery disease (CD) in middle aged people has decreased by 31% during the last decade. However, atherosclerotic cardiovascular disease continues to be the most frequent cause of death, both in men and women. The general mortality rate due to cardiovascular diseases is 235.5 per 100,000 population and CD only caused 1 out of 6 deaths in the United States in 2010 [1].
Familial hypercholesterolemia (FH) is an autosomal dominant disease characterized by very high serum cholesterol and low density lipoprotein levels, being clinically expressed intendinous xanthomas and premature atherosclerosis [2]. More than 85% of FH cases are attributable to inherited mutations of the LDL receptor gene (LDLR), of which more than 1,600 mutations have been identified [3]. The LDLR gene mutation leads to defects in LDLc capture in the blood. Less frequently, FH may also be caused by mutations in the proprotein convertase subtilisin/kexintype 9 (PCSK9) gene, or other rare mutations in related genes [4, 5].
Heterzygous FH is the most common form of the disease (prevalence of approximately 1 in 300 to 500 individuals worldwide, and which can be as high as 1 in 100 individuals in some special populations) [3-6]. Homozygous autosomal dominant FH is a very rare form of the disease (prevalence of 1 in 1 million individuals). Some populations, such as French Canadians, Ashkenazi Jews, Lebanese and Dutch of African origin, are at a higher risk for FH due to the increased prevalence of mutations associated with heterozygous FH [7-10].
FH is characterized by severe hypercholesterolemia which results in premature cardiovascular disease. Individuals with FH have a greater risk of cardiac events such as myocardial infarction and death due to premature CD, especially in patients with severe forms of the disease, if they are not treated [11, 12]. Homozygous FH is a serious and aggressive form of the disease which frequently does not respond to traditional hypercholesterolemia treatment due to the lack of functional LDL-c receptors [12, 13]. In general, the average age at diagnosis of cardiovascular manifestations is 20 years [12]. The elevation of LDL-c levels reflects the severity of the genetic mutation. Patients with heterozygous FH typically present levels double or triple those of healthy individuals (approximately 200-400mg/dL); while patients with homozygous FH have LDL-c levels that may be more than 6-10 times normal values (> 600 mg/dL) [13].
Homozygous FH is associated with coronary artery disease and premature death, with several reports of individuals under 17 years of age who developed severe coronary artery stenosis [2, 14] and supravalvular aortic stenosis [15], an increase in intima-media thickness of the carotid and femoral arteries [16], and tendinous xanthomas caused by cholesterol deposits on tendons and skin, observed mainly on the elbows, knees, Achilles heel, and dorsum of hands and feet. Tendinous xanthomas are highly suggestive of homozygous FH and are a basic criterion in the clinical diagnosis of this disease. Xanthomas are seen in up to 50% of patients with FH [17]. Excess cholesterol may also be deposited on the cornea, causing pigmentation manifested as a corneal arc.
Patients and Methods
We describe our experience with two young patients who presented to our Interventional Cardiology department in September and October, 2013, with clinical characteristics of homozygous FH and severe CD, and underwent percutaneous myocardial revascularization with drug-eluting stent (DES) implantation.
Case 1
Fifteen year old male (Tables 1 and 2). Presents with a two week history of chest pain and decreased functional class. Clinical presentation on admission was recorded as myocardial infarction without ST elevation, based on positive troponin and electrical changes in the anterior wall. He has a history of homozygous FH (Figure 1) with cutaneous xanthomas (Figure 2), severe hyperlipidemias (LDL-c up to 850 mg/dL), grade 1 arterial hypertension, Ebstein´s Anomaly and WPW syndrome which was ablated in September, 2011. Due to FH and poor control of LDL-c levels he underwent liver transplant in June, 2013. Submaximal stress test was positive for ischemia. Currently being treated pharmacologically with ezetimibe 10mg/day, simvastatin 20mg/day, amlodipine 5mg/day, prednisolone 2.5mg/day and tacrolimus 3mg/ day. Coronary angiography shows severe CD in the proximal anterior descending artery and the ostial right coronary. He underwent coronary angioplasty with implantation of 2.25x15mm and 3.0x12mm Resolute IntegrityTM (Medtronic Vascular, Santa Rosa, CA) drug-eluting stents, respectively, post-dilation with high pressure non-compliant balloon at 18 atms. (Figures 3 and 4). He was asymptomatic at six month clinical follow-up.
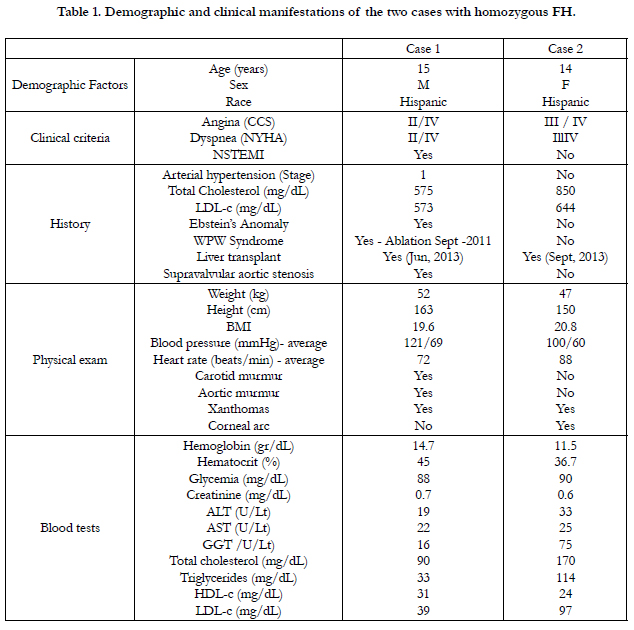

Table 1. Demographic and clinical manifestations of the two cases with homozygous FH.
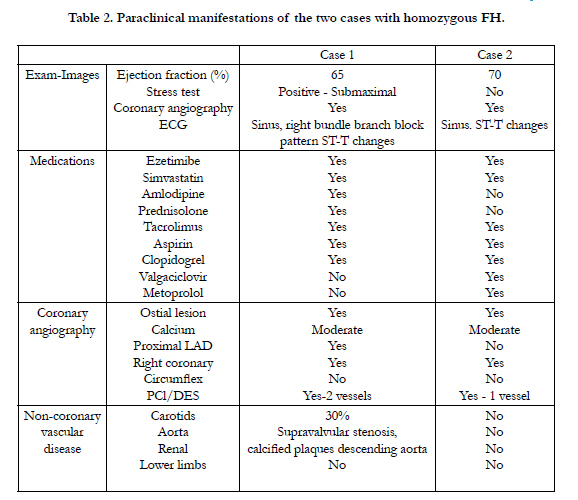
Table 2. Paraclinical manifestations of the two cases with homozygous FH.
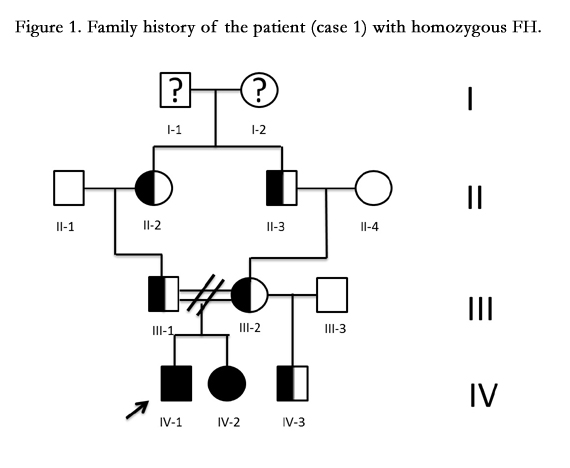
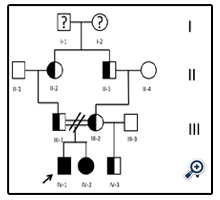
Figure 1. Family history of the patient (case 1) with homozygous FH.
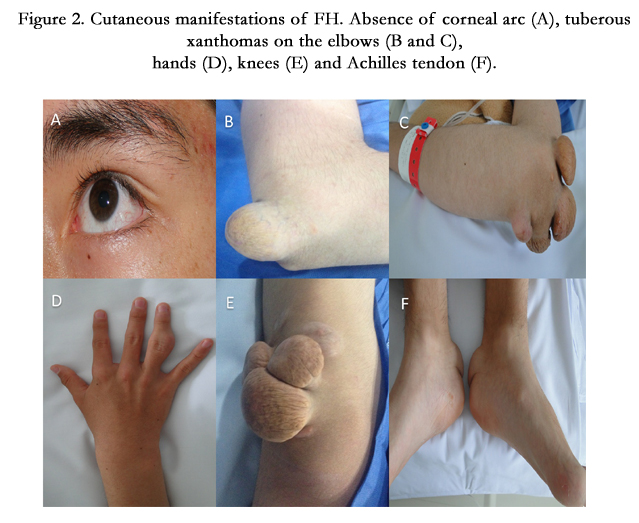
Figure 2. Cutaneous manifestations of FH. Absence of corneal arc (A), tuberous xanthomas on the elbows (B and C), hands (D), knees (E) and Achilles tendon (F).
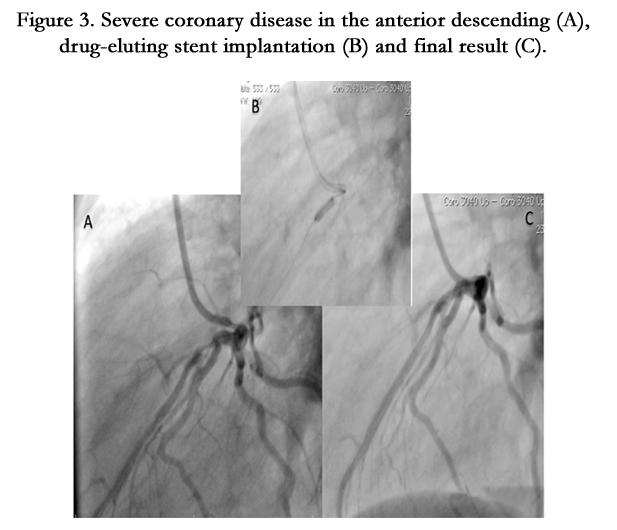
Figure 3. Severe coronary disease in the anterior descending (A), drug-eluting stent implantation (B) and final result (C).
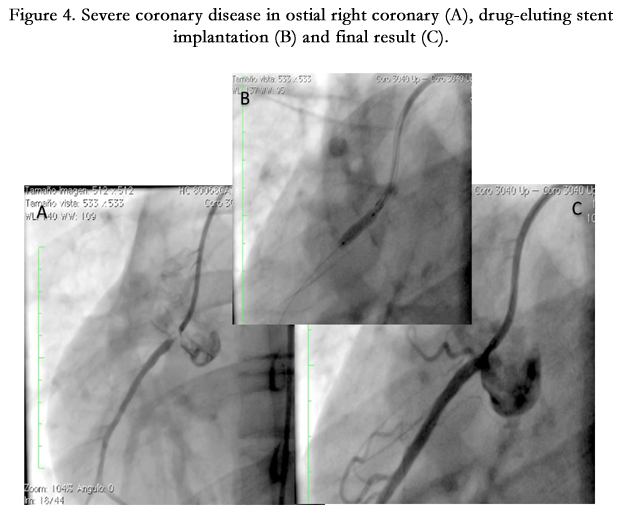
Figure 4. Severe coronary disease in ostial right coronary (A), drug-eluting stent implantation (B) and final result (C).
Case 2
Fourteen year old female (Tables 1 and 2). Recent onset of chest pain and dyspnea. History of homozygous FH (Figure 5). A review of the family history shows an extensive history of heterozygous FH and cardiovascular events in both maternal and paternal families. Genetic analysis through LDLR and ApoB gene sequencing showed a homozygous or hemizygous mutation onan LDLR intron and a homozygous or hemizygous mutation on an LDLR exon. She presents with cutaneous xanthomas and corneal arc (Figure 6), severe hyperlipidemia (LDL-c up to 610mg/dL) and liver transplant in September, 2013. Currently being treated with ezetimibe 10mg/day, simvastatin 20mg/day, valganciclovir, tacrolimus 3mg/day and ursacol. Coronary angiography showed severe CD in ostial right coronary. Coronary angioplasty was carried out, implanting a 2.5x15 mm Resolute IntegrityTM (Medtronic Vascular, Santa Rosa, CA) drug-eluting stent following dilation with a high pressure non-compliant balloon at 16 atms. (Figure 7). She was asymptomatic at six month clinical follow-up.
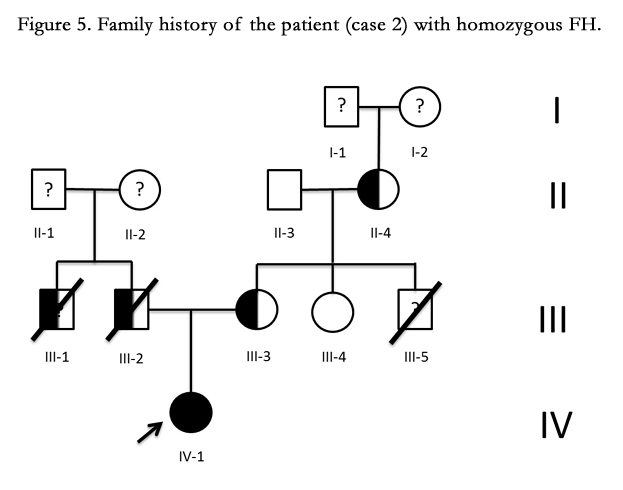
Figure 5. Family history of the patient (case 2) with homozygous FH.
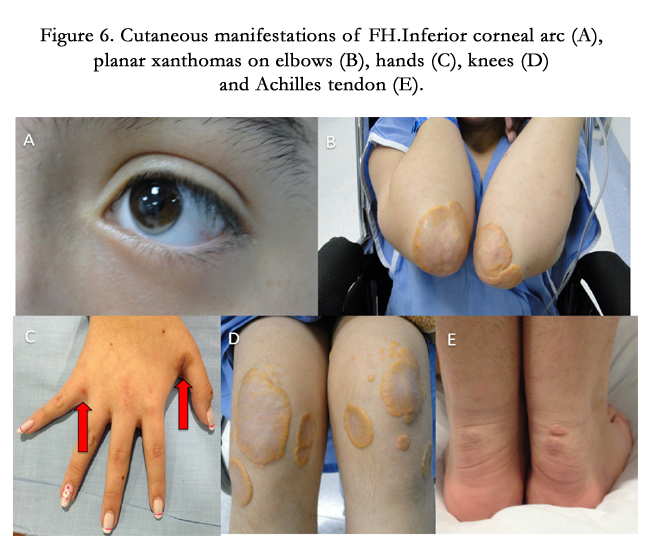
Figure 6. Cutaneous manifestations of FH.Inferior corneal arc (A), planar xanthomas on elbows (B), hands (C), knees (D) and Achilles tendon (E).
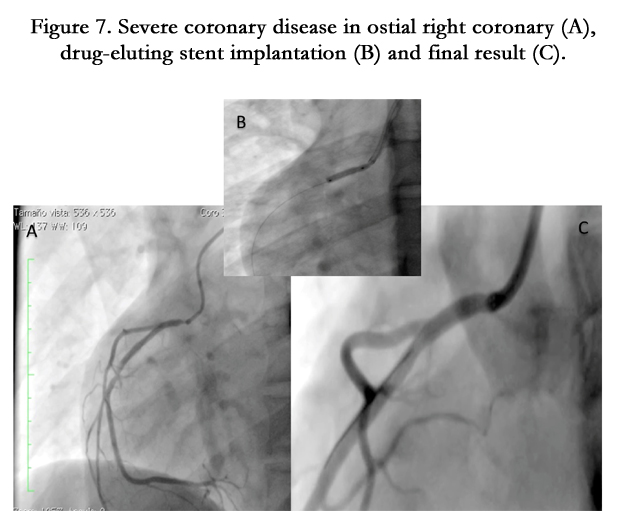
Figure 7. Severe coronary disease in ostial right coronary (A), drug-eluting stent implantation (B) and final result (C).
Discussion
FH is a monogenic, autosomal dominant disorder caused by mutations in the LDL receptor gene. We present two cases of homozygous FH in young people 14 and 15 years of age and severe CD of main vessels (anterior descending and right coronary) who underwent successful DES implantation. They have the particular characteristic of cardiovascular manifestations (chest pain) at their age, and being post-liver transplant (3 months and 2 months, respectively) with no evidence of rejection and a normal lipid profile.
The literature describes a mean age for cardiovascular disease diagnosis in patients with homozygous FH of 20 years [11, 18]. Several publications have been issued with homozygous patients under 17 years of age who developed severe CD and supravalvular aortic stenosis in the ´80s when percutaneous coronary revascularization with stents was just beginning [2, 15, 19, 20].
Tellez A et al. [28] described in 2010 the vascular response which occurs following implantation of a bare metal stent (BMS) in a porcine model with FH, showing a more agressive neointimal formation than that which occurs in domestic animals, which suggests the need to consider the use of DES in FH conditions. Recently, Shankarappa RK et al. [14] reported 5 patients treated at the Sri Jayadeva Cardiology Institute in Bangalore, India. Four men and one woman. They all presented with coronary symptoms, tendinous xanthomas and severe CD. All had total cholesterol levels >435mg/dL and LDL-c >392mg/dL. Arterial revascularization surgery was necessary in 3 patients, and the other two required angioplasty with successful stent implantation. In one 24 year old man, a drug-eluting stent in the anterior descending and a bare metal stent in the circumflex, and in a 17 year old woman a drug-eluting stent in the left main trunk.
Percutaneous coronary intervention (PCI) is commonly used in adult patients with severe CD. In patients with unstable angina or NSTEMI, an early invasive strategy (that is, angiogiographic diagnosis with the intention of performing a revascularization) is indicated with Class I and B level of evidence [27]. But PCI information in children and adolescents is very limited. Most cases described in those under 18 years of age have had a non-atherosclerotic indication such as transplant coronary vasculopathy [21, 23, 25, 26], coronary dissection [21], and coronary compression or occlusion as a sequela of prior heart surgery [21, 22, 24].
The Guidelines published by the US National Lipid Association (NLA) and NICE in the United Kingdom recommend a reduction in LDL-c concentration of >50% with respect to pre-treatment levels in patients with FH [34, 35,36, 37]. The Canadian and European guidelines recommend a reduction of LDL-c levels to <116mg/dL in patients at moderate risk for cardiovascular disease; <97mg/dL in patients at high risk, and <70mg/dL in patients at very high risk. There are different treatment options already available for patients with FH. Diet, pharmacologic therapy, lipid apheresis, and some surgical techniques, such as portacaval shunt surgery which limits the absorption of cholesterol and promotes the loss of bile acids, and liver transplant are among the different treatments. Liver transplant to make functional LDLRs available is an alternative for the most serious cases [29].
A few years after Brown and Goldstein won the Nobel Prize in Medicine in 1985 when they described that FH was due to an intrinsic defect of the hepatocyte in the deficiency of LDL-c receptors as an explanation for the extremely high serum cholesterol levels [20], Starzl et al. [33] reported the first orthotopic liver transplant performed for reasons beyond the usual indication of liver failure, as treatment for hyperlipidemia in the context of homozygous FH. It was the first time an anatomically normal liver was surgically removed from a six year old patient, for presumptive treatment of a hypercholesterolemic condition.
As it is known that most LDL receptors are found in the liver, liver transplant has become the treatment of choice for affected patients who do not respond to routine pharmacologic treatments [30, 31]. The transplanted liver retains the specific qualities of the donor and therefore the transplant can be a source of abundant functional LDL receptors, and may lead to a cure of the hypercholesterolemia [29]. Progress in experience of the transplant groups, long-term immunosuppression, improvements in surgical techniques and immunologic methods have yielded favorable results following transplant both in the adult and the pediatric populations [32, 33].
Conclusions and Recommendations
FH is an autosomal dominant genetic disorder associated with elevated LDL-c levels, which may lead to premature cardiovascular disease. The early diagnosis of FH is important in order to prevent morbidity and mortality. FH is generally diagnosed based on clinical characteristics, family history, and serum cholesterol levels. The current Guidelines highlight the importance of lowering LDL-c levels in patients with FH. We report two patients with homozygous FH, in young people 14 and 15 years old, with clinical manifestations and severe hypercholesterolemia treated pharmacologically and with liver transplant. They presented acute coronary syndrome without ST elevation and severe CD treated successfully with implantation of drug-eluting stents.
References
- Alan S. Go, Mozaffarian D, Véronique L. Roger, Emelia J. Benjamin, Jarett D. Berry, et al. (2014) on behalf of the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart Disease and Stroke Statistics—2014 Update A Report From the American Heart Association. Circulation 129: 399-410.
- Sprecher DL, Schaefer EJ, Kent KM, Gregg RE, Zech LA, et al. (1984) Cardiovascular features of homozygous familial hypercholesterolemia: analysis of 16 patients. Am J Cardiol 54: 20–30.
- Hopkins PN, Toth PP, Ballantyne CM, Rader DJ (2011) Familial hypercholesterolemias: prevalence, genetics, diagnosis and screening recommendations from the National Lipid Association Expert Panel on Familial Hypercholesterolemia. J ClinLipidol 5(3):9-17.
- Liyanage KE, Burnett JR, Hooper AJ, van Bockxmeer FM (2011) Familial hypercholesterolemia: epidemiology, Neolithic origins and modern geographic distribution. Crit Rev Clin Lab Sci 48(1):1-18.
- Goldberg AC, Hopkins PN, Toth PP, Christie M. Ballantyne, Daniel J. Rader, et al. (2011) Familial hypercholesterolemia: screening, diagnosis and management of pediatric and adult patients: clinical guidance from the National Lipid Association Expert Panel on Familial Hypercholesterolemia. J ClinLipidol 5(3):1-8.
- Wierzbicki AS, Humphries SE, Minhas R (2008) Familial hypercholesterolaemia: summary of NICE guidance. BMJ 337:1095.
- Hobbs HH, Brown MS, Russell DW, Davignon J, Goldstein JL (1987) Deletion in the gene for the low-density-lipoprotein receptor in a majority of French Canadians with familial hypercholesterolemia.N Engl J Med 317(12): 734-7.
- Moorjani S, Roy M, Gagne C, Davignon J, Brun D, Toussaint M, Lupien P (1989) Homozygous familial hypercholesterolemia among French Canadians in Québec province. Arteriosclerosis, Thrombosis, and Vascular Biology 9(2): 211-216.
- Yuan G, Wang J, Hegele RA (2006) Heterozygous familial hypercholesterolemia: an underrecognized cause of early cardiovascular disease. CMAJ 174(8): 1124-9.
- Zeegers MP, van Poppel F, Vlietinck R, Spruijt L, Ostrer H (2004) Founder mutations among the Dutch. Eur J Hum Genet 12(7): 591-600.
- Kolansky DM, Cuchel M, Clark BJ, Paridon S, McCrindle BW (2008) Longitudinal evaluation and assessment of cardiovascular disease in patients with homozygous familial hypercholesterolemia. The American journal of cardiology 102(11): 1438-1443.
- Naoumova RP, Thompson GR, Soutar AK (2004) Current management of severe homozygous hyper cholesterolaemias. CurrOpinLipidol 15(4): 413-22.
- Liyanage KE, Hooper AJ, Defesche JC, Burnett JR, van Bockxmeer FM (2008) High-resolution melting analysis for detection of familial liganddefective apolipoprotein B-100 mutations. Ann ClinBiochem 45(2): 170-6.
- Shankarappa RK, Moorthy N, Bhat SPS, Dwarakaprasad R, Nanjappa MC (2009) The challenge produced by familial homozygous hypercholesterolemia when treating premature coronary arterial disease in the Young. Cardiol Young 19: 257–63.
- Allen JM, Thompson GR, Myant NB, Steiner R, Oakley CM (1980) Cardiovascular complications of homozygous familial hyper-cholesterolemia. Br Heart J 44: 361–8.
- Hutter CM, Austin MA, Humphries SE (2004) Familial hypercholesterolemia, peripheral arterial disease, and stroke: a HuGEminireview. Am J Epidemiol 160(5): 430–5.
- Civeira F, Castillo S, Alonso R, Meriño-Ibarra E, Cenarro A, et al. (2005) Tendon xanthomas in familial hypercholesterolemia are associated with cardiovascular risk independently of the low-density lipoprotein receptor gene mutation. Arteriosclerosis, thrombosis, and vascular biology 25(9): 1960-1965.
- Robinson JG (2013) Management of Familial Hypercholesterolemia:A Review of the Recommendations from the National Lipid Association Expert Panel on Familial Hypercholesterolemia. J Manag Care Pharm 19(2): 139-49.
- Mabuchi H, Koizumi J, Shimizu M, Takeda, R. (1989) Development of coronary heart disease in familial hypercholesterolemia. Circulation 79(2): 225-232.
- Goldstein JL, Brown MS (1983) Familial hypercholesterolemia. In: The metabolic basis of inherited disease (5th edtn) Stanbury JB, Wyngaarden JB, Fredrickson DS, Goldstein JL, Brown MS, editors. McGraw-Hill, New York, NY 672-712.
- Schneider AE, Johnson JN, Taggart NW, Cabalka AK, et al. Percutaneous coronary intervention in pediatric and adolescent patients. Congenit Heart Dis. 2013; Aug 15:1-7.
- Natal-Hernandez L, Meadows J, Shunk KA, Boyle AJ (2013) Percutaneous retrograde recanalization of a chronic total coronary artery occlusion in a 7 year old.Cardiovascular Revascularization Medicine 14: 113–7.
- Lee MS, Sachdeva R, Hyun Kim M, Sachdeva R (2012) Long-term outcomes of percutaneous coronary intervention in transplant coronary artery disease in pediatric heart transplant recipients. J Invasive Cardiol 24(6): 278-81.
- Kaichi S,Doi H, Tsurumi F, Heike T (2011) Long-term outcome of sirolimus- eluting stent implantation for left main coronary artery stenosis in infancy. Pediatr Cardiol 32: 94–7.
- Sachdeva R, Seib PM, Frazier EA, Sachdeva R (2009) Percutaneous coronary intervention using drug-eluting stents in pediatric heart transplant recipients. Pediatr Transplantation 13:1014–9.
- Salloum JG, Dodd DA, Slosky D, Zhao DXM (2007) Treatment of unprotected left main coronary artery stenosisin a 5-year-old heart transplant patient using asirolimus-eluting stent. J Heart Lung Transplant 26: 1061–4.
- Levine GN, Bates ER, Blankenship JC, Bailey SR, Bittl JA, et al. (2011) ACCF/AHA/SCAI guideline for percutaneous coronary intervention: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines and the Society for Cardiovascular Angiography and Interventions.Journal of the American College of Cardiology 58(24): 44-122.
- Tellez A, Krueger CG, Seifert P, Winsor-Hines D, Piedrahita C, et al. (2010) Coronary bare metal stent implantation in homozygous LDL receptor deficient swine induces a neointimal formation pattern similar to humans. Atherosclerosis 213(2): 518-524.
- Nemati MH, Astaneh B (2010) Optimal management of familial hypercholesterolemia: Treatment and management strategies. Vasc Health and Risk Management (6): 1079–88.
- Moghadasian MH, Frohlich JJ, Saleem M, Hong JM, Qayumi K, et al. (2001) Surgicalmanagement of dyslipidemia: clinical and experimental evidence. J Invest Surg 14(2): 71-8.
- Bilheimer DW, Goldstein JL, Grundy SM, Starzl TE, Brown MS (1984) Liver transplantation to provide low-density-lipoprotein receptors and lower plasma cholesterol in a child with homozygous familial hypercholesterolemia. N Engl J Med 311(26): 1658–64.
- Lopez-Santamaria M, Migliazza L, Gamez M, Murcia J, Diaz-Gonzalez M, et al. (2000) Liver transplantation in patients with homozygotic familial hypercholesterolemia previously treated by end-to-side portocaval shunt and ileal bypass. Journal of pediatric surgery 35(4): 630-633.
- Maiorana A, Nobili V, Calandra S, Francalanci P, Bernabei S, (2011) Preemptive liver transplantation in a child with familial hypercholesterolemia.PediatrTransplantation 15: 25–29.
- Goldberg AC, Hopkins PN, Toth PP, Ballantyne CM, Rader DJ, et al. (2011) Familial hypercholesterolemia: screening, diagnosis and management of pediatric and adult patients: clinical guidance from the National Lipid Association Expert Panel on Familial Hypercholesterolemia. J ClinLipidol 5: 133–40.
- Reiner Z, Catapano AL, De Backer G, Graham I, Taskinen MR, et al. (2011) ESC/EAS Guidelines for the management of dyslipidaemias: the Task Force for the management of dyslipidaemias of the European Society of Cardiology (ESC) and the European Atherosclerosis Society (EAS). Eur Heart J 32:1769–1818.
- Grundy SM, Cleeman JI, Merz CN, Brewer HB Jr, Clark LT, et al. (2004) Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III Guide- lines. J Am CollCardiol 44:720–32.
- Genest J, McPherson R, Frohlich J, Anderson T, Campbell N, et al. (2009) Canadian Cardiovascular Society/Canadian guidelines for the diagnosis and treatment of dyslipidemia and prevention of cardiovascular disease in the adult – 2009 recommendations. Can J Cardiol 25: 567–9.





