Apoptosis Induction by Cestrum parqui L’Hér. leaves on HL-60 Cell Line: Identification of Active Phytomolecules
Chenni H1*, Torres JL2, Estévez F3, Ali KM4, Ghosh D4, Trabelsi M1
1. Laboratory of Histology and Embryology, Faculty of Medicine, 4002 Sousse, Tunisia.
2. Institute for Advanced Chemistry of Catalonia IQAC-CSIC, Jordi Girona 18-26 08034- Barcelona.
3. Dept. of Biochemistry, Molecular Biology and Physiology, University of Las Palmas de Gran Canaria, Plaza Dr. Pasteur s/n, 35016 Las Palmas de
Gran Canaria, Spain.
4. Dept. of Bio-Medical Laboratory Science and Management (U.G.C Innovative Funded Department), Vidyasagar University, Midnapore-721 102, West
Bengal, India.
*Corresponding Author
Hanene Chenni,
Faculty of Medicine,
4002, Sousse, Tunisia.
Tel: +216 73 222 600
Fax: +216 73224899
E-mail: chenni_han@yahoo.fr
Article Type: Research Article
Received: March 19, 2015; Accepted: April 18, 2015; Published: April 22, 2015
Citation: Chenni H, et al.,(2015) Apoptosis Induction by Cestrum parqui L’Hér. leaves on HL-60 Cell Line: Identification of Active Phytomolecules. Int J Cancer Stud Res. S1:001, 1-8. doi: dx.doi.org/10.19070/2167-9118-SI01001
Copyright: Chenni H© 2015. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
Abstract
Background: In our time, there is an urgent need to discover natural anticancer products without toxic side’s effects.
Objective: The present study was conducted to investigate the anticancer activity of the methanolic extract of Cestrum parqui leaves as well as identification of the active compounds in this concern.
Materials and Methods: Eight column chromatographic fractions were tested against human leukaemia cell line (HL-60) and the cell viability was measured by dimethylthiazol tetrazolium assay. Studies of apoptosis, cell cycle and DNA fragmentation were carried out following the standard protocol. Effective fractions were analysed by high performance liquid chromatography-ultra violet, HPLC-evaporative light scattering detector and preparative-HPLC. Finally the active compounds were identified by gas chromatography coupled to mass spectrometry.
Results: Out of eight, fraction II showed remarkable inhibition on the growth of HL-60 cell line and fraction I showed moderate activity on such inhibition. Mechanisms of the inhibition were identified as cell cycle arrestation and apoptosis induction by the said fractions. Chromatographic and mass spectrometric analysis of the fraction II indicated the presence of oleanolic and ursolic acids as biologically active compounds.
Conclusion: This study is the first investigation that shows Cestrum parqui has anticancer activity and the responsible compounds are oleanolic and ursolic acids which may be used for chemotherapy of certain cancer in future after extensive work in this line.
2.Introduction
3.Methods and Materials
3.Preparation of the methanolic extract
4.Bioactivity Study
4.1 Cell culture and treatments
4.2 Cell proliferation analysis
4.3 Cell apoptosis rate determination by Quantitative Nuclear Fluorescence Morphology (QNFM) assay
4.4 Cell cycle analysis
4.5 DNA fragmentation analysis
5.Instrumental Analysis
5.1.Analytical HPLC
5.2.LC-PDA-ELSD analysis
5.3.Prep-HPLC
5.4.Derivatization of purified compound
5.4.GC-MS conditions
5.5.Statistical analysis
6.Results
6.1.Cell proliferation analysis
6.2.Cell apoptosis rate determination by QNFM assay
6.3.Cell cycle analysis
6.4.DNA fragmentation analysis
6.5.HPLC-PDA–ELSD
6.5.Identification by GC-M
7.Discussion
8.Conclusion
9.Acknowledgement
10.References
Keywords
Anticancer Activity; Ursolic and Oleanolic Acids; HPLC-UV; HPLC-ELSD; GC-MS.
Introduction
Cancer, one of the leading causes of human death, has attracted more attention due to the complexity of etiology, diagnosis and treatment [1]. In recent years, chemotherapeutic management against various types of cancer including leukemia has been increased rapidly though some times cancer cells resistance against chemotherapy [2, 3]. In this respect, search for effective anticancer agent that inhibit the proliferation of the cells as well as inhibit the resistance nature of the cancer cells and induce differentiation of the monocytes is an ongoing challenge. Therefore to find out anticancer agent with multi-target efficacies, attention has been focused on plant products as the plant products have been used as remedies for several human diseases including cancer from ancient times with lower side effects [4]. The reasons for using them that they contain variety of chemical compounds usually secondary metabolites with therapeutic values [5]. Cestrum parqui is belonging to the family Solanaceae, usually known as “green or Chilean cestrum” in Tunisia, are rich in various secondary metabolites such as saponins, polyhydroxylated terpenes (C13 norisoprenoids, sesquiterpenes, spirostanes and pseudosapogenins) and ent-kaurene glycosides (parquin and carboxyparquin) [6-8]. Several scientific reports demonstrate that C. parqui exhibits a wide spectrum of pharmacological activity when administered systemically or in isolated organ preparations. For example, it is used to treat headache, rheumatic pain, scabies, dermatosis, diarrhaea and metrorraghia [7]. The plant is also used in Chilean folk medicine as antifebrile and for the treatment of fever and inflammation [8] though there is no information about the anticancer efficacy of this plant.
In the present study, anticancer activity of leaves of C. parqui was investigated on human myeloid leukaemia cell line (HL-60). For this purpose, first methanol extract of C. parqui leaves was fractioned by column chromatography followed by all the fractions were examined on cell line (HL-60). Simultaneously an attempt was taken to identify the biologically active compounds from the effective fraction responsible for anticancer activity.
Methods and Materials
The leaves of C. parqui were collected from the National Institute of Agronomy Garden, Tunisia. Collected seeds were dried in a steam room at 40°C for 3 days followed by ground into powder form. The powdered material was extracted in methanol for 7 days and it was repeated for three times. Then the collected methanolic phases were combined and the residue was removed by filtration. The filtrate was concentrated under reduced pressure by rotary evaporator to obtain methnolic Extract (ME).This extract was subjected to column chromatography using silica gel as the stationary phase and eluted by solvents as per increasing polarity i.e. CH2Cl2-MeOH (20:1→0:1). Seventy fractions (100mL each) were collected and based on their thin layer chromatographic (TLC) profiles fractions were combined and ultimately eight pooled fractions (F-I to F-VIII) were obtained. Finally, entire pooled fraction was concentrated separately under reduced pressure and the residue was used for bioactivity study.
Bioactivity Study
HL-60 cell line was cultured in RPMI-1640 medium containing 10% (v/v) heat-inactivated fetal bovine serum, 100units/mL penicillin and 100μg/mL streptomycin under 5% CO2-95% air humidified incubator at 37°C. The cell numbers were counted using Neubauer’s counting chamber and the viability was determined by trypan blue (0.025%) exclusion method.
Dilutions of each fraction were performed just before used in the culture media using 0.5 % dimethyl sulfoxide (DMSO). The cells were treated in triplicate manner with various concentrations of each fraction. In control, only 0.5 % DMSO was used as the said concentration is non toxic to the cells.
Analysis of cell proliferation in fraction treated as well as in control cells was carried out by 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyl tetrazolium bromide (MTT) assay [9].
The morphological changes in the cells were assessed by Hoechst staining under fluorescent microscope. For this purpose, 5x105 HL-60 cells/well were plated in 6 well plates. After 24h of treatment with 30μg/mL of fraction II, the cells were harvested and fixed in 3% paraformaldehyde followed by incubation at room temperature for 10 min. The cells were washed in phosphate buffer saline (PBS) and incubated for 15 min at room temperature in 50μL of PBS containing 20μg/mL bis-benzimide trihydrochloride (Hoechst 33258).
Finally, 10 μL aliquots of the cells were placed on glass slides and 500 cells were counted and scored for the incidence of apoptotic chromatin condensation in triplicate samples under Zeiss fluorescent microscope.
Alteration of the cell DNA content, histogram measurements of hypodiploid DNA formation was carried out by flow cytometer using EPICS™ cytometer-Beckman Coulter [10].
In brief, cells were treated with 30μg/mL of fraction II for 24 h followed by harvested, washed and fixed with ice-cold 75 % ethanol for 1h. The cells were centrifuged, pellets were suspended in PBS and treated with RNase A at the concentration of 100 μg/mL. Propidium iodide at the concentration of 50μg/mL was added to the sample and kept in dark for 30 min to stain DNA. For each determination 10, 000 cells were counted and the cells with decreased DNA staining composed of apoptotic cells were expressed in terms of percentage (%). Histograms were analyzed with the Expo 32 ADC Software™.
The isolation of fragmented DNA was carried out according to standard protocol [9]. Cells were harvested and washed with PBS and pelleted by centrifugation. Then the pellets were treated with 30μL of lysis buffer (50 mM Tris-HCl, pH 8.0) containing 10 mM EDTA, 0.5% SDS, 1 μg/μL RNase (Sigma) for 1 h at 37°C. Finally 3μL of proteinase K (10μg/μL) was added and the mixture was incubated for 1h at 50°C.
DNA was extracted with 150μL of phenol-chloroform-isoamyl alcohol (24:24:1) and dissolved in loading buffer (10mM EDTA, pH 8.0, 40% sucrose and 0.25% bromophenol blue). Aliquot was loaded on 1.8% agarose gel, stained with ethidium bromide and the expression of DNA fragments were observed under UV illumination.
Instrumental Analysis
The fractions were analyzed by high performance liquid chromatography with diode array (HPLC-DAD) using a Hitachi Lachrom Elite HPLC system (San Jose, CA USA) equipped with a quaternary pump, auto sampler, in-line degassing unit, temperature control unit, photo-diode array UV detector and fitted with an analytical column Kromasil C18 (Teknokroma, Barcelona, Spain) (25 × 0.4 cm i.d., 100-Å, 5-μm particle size). Acquisitions were made using EZChrom Elite 3.1.3 from Scientific Software Inc. (Pleasanton, CA, USA). Load: 40 μL, 10 μg. Elution: [A] 0.1% (v/v) aqueous TFA, [B] 0.08% (v/v) TFA in water/CH3CN 1:4, gradient 12 to 30 % [B] over 30 min at the flow rate of 1mL/min. DAD detection was performed from at 210 to 380 nm. Data were acquired in triplicate.
The HPLC work was performed on an Alliance 2695 apparatus coupled with a 996 UV diode array detector (Waters Corporation, Milford, MA, USA) together with Polymer Laboratories (Amherst, MA, USA) PL-ELS 1000 evaporative light scattering (ELS) detector. Fractions were collected on a Waters fraction collector III (Waters Corporation). A column Kromasil C18 (Teknokroma, Barcelona, Spain) (25 × 0.4 cm i.d., 100-Å, 5-μm particle size) was used as stationary phase. The mobile phase consist of water (0.1% acetic acid) (A) and methanol (B) at the flow rate of 1 mL/min and the injection volume was 100 μL. Analysis was carried out using the following gradient elution: 0-20min:80%B, 20-40min:100% B. The method of ELS detector was set up as follows: Gas flow was 1.5 L/min, Nebulizer temperature: 90°C, Evap. temperature: 120°C and the detection wavelength was 254 nm.
Preparative RP-HPLC was performed on a Waters Prep LC pumping system (Milford, MA) equipped with Waters 2489 UV/V is detector and X-Terra Prep MS C18 column (Waters). Separation was done using the same gradient elution as in HPLC-PDAELSD conditions. Flow rate was 10mL/min, detection wavelength 254 nm and injection volume was 20μL.
The derivatization of the compound was carried out according to standard method [11]. In brief, 0.2μg of purified compounds were derivatized by adding 100μL of silylation reagent N-methyl-N-trimethyl silyl tri-fluoro acetamide (MSTFA) and 50μL of pyridine. The reaction mixture was kept for 2 h at 70°C. After removing the solvents, 5μL of tetrahydrofuran (THF) was added followed by 1μL of the solution was injected into the gas chromatography coupled to mass spectrometry (GC-MS).
GC-MS analysis was carried out using BD-5 capillary column where flow rate was 1mL/min. The system was operated under the following conditions: injection temperature, 290°C; transfer line temperature, 300°C; oven temperature program, 60(l), and rate 12°C/min; final temperature, 320°C. The mass spectrometer was monitored to scan 40-800 uma with an ionizing voltage at 70eV.
Statistical comparison between the groups was done by student’s t test using software STATISTICA. Differences between the groups were considered significant when p<0.05.
Results
Effect of different fractions (I-VIII) on the viability of HL-60 cells was measured by MTT assay. After 72h, the viability of HL-60 cells was reduced more than 50% when HL-60 cells were treated with fractions (I-IV) at the dose of 100μg/mL. Though, the cell viability was increased more than 100% when the cells were treated with fractions (V-VIII). This data suggested that only fractions (I-IV) were effective in the inhibition of HL-60 cell proliferation (Figure1A).
According to the National Cancer Institute (NCI), plant extracts and pure compounds with cytotoxic ED50 (Effective Dose 50) values ≤30μg/mL and ≤40μg/mL, respectively, are considered active. [12] Therefore, further MTT test was carried out using the fractions I-IV at lower dose i.e. 30 μg/mL to search out the threshold dose in this concern. The result indicated that the fractions II was most active whereas fractions (I, III and IV) showed moderate activity in this purpose (Figure1B). Result focused that fraction III at the dose of 100μg/mL is more efficient than Fraction II (Figure 1A). But, this is not applicable at the dose of 30μg/mL (Figure 1B) which may be the difference in composition of these two fractions.
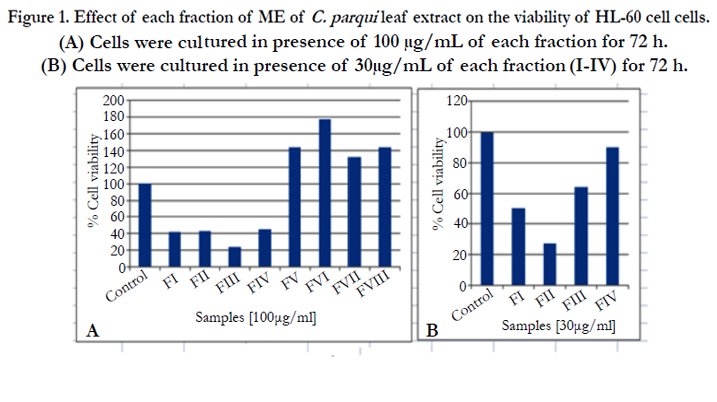
Figure 1. Effect of each fraction of ME of C. parqui leaf extract on the viability of HL-60 cell cells. (A) Cells were cultured in presence of 100 μg/mL of each fraction for 72 h. (B) Cells were cultured in presence of 30μg/mL of each fraction (I-IV) for 72 h.
For the determination of apoptosis, HL-60 cells were treated with 30μg/mL of each active fraction (I, II and III) and analyzed by light microscopy. Fraction II treated cells showed the presence of more number of apoptotic cells in respect to control (Figure 2, Figure 2A control and Figure 2B cells treated with 30μg/mL of fraction II) which also supports the results obtained by MTT test. So, the results obtained by MTT test and light microscopy analysis indicated that fraction II is the most potent to induce apoptotic cell death.
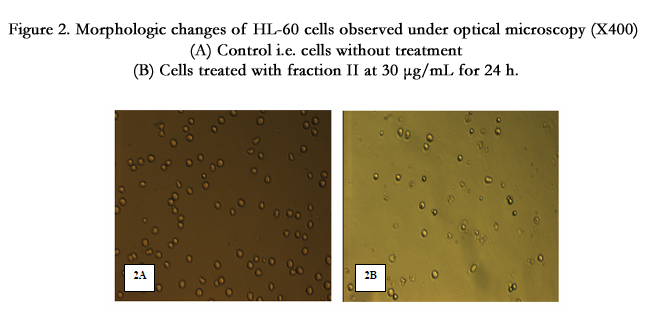

Figure 2. Morphologic changes of HL-60 cells observed under optical microscopy (X400) (A) Control i.e. cells without treatment (B) Cells treated with fraction II at 30 μg/mL for 24 h.
Fluorescence microscopic observation after Hoechst 33258 staining of fraction II treated HL-60 cells showed that the control cells were uniformly blue and the apoptotic cells were blue having bright blue dots in their nuclei, representing the apoptotic bodies (nuclear fragmentation) (Figure 3A). Indeed, fraction II resulted dose-dependent increase in apoptotic-death of cells during 24 h (Figure 3B).
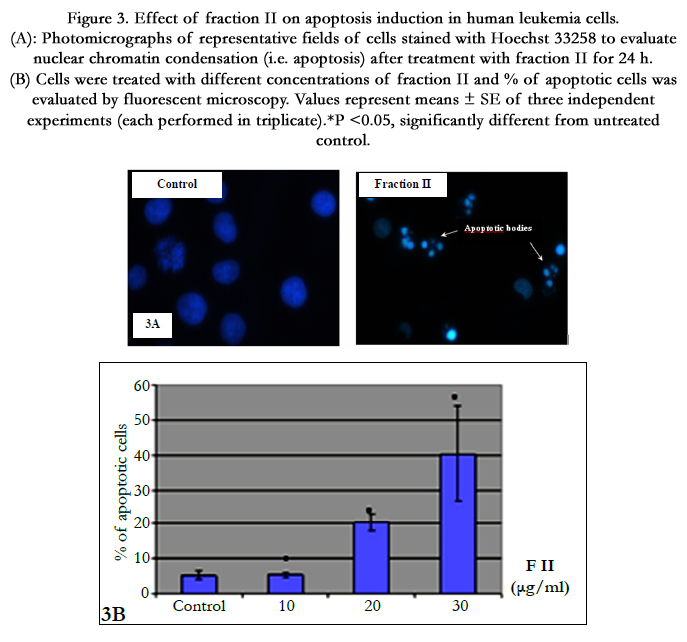
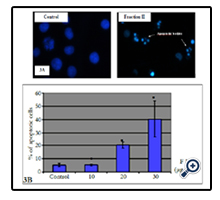
Figure 3. Effect of fraction II on apoptosis induction in human leukemia cells. (A): Photomicrographs of representative fields of cells stained with Hoechst 33258 to evaluate nuclear chromatin condensation (i.e. apoptosis) after treatment with fraction II for 24 h. (B) Cells were treated with different concentrations of fraction II and % of apoptotic cells was evaluated by fluorescent microscopy. Values represent means ± SE of three independent experiments (each performed in triplicate). *P <0.05, significantly different from untreated control.
To explain the observed inhibition in proliferation, cell cycle distribution after 24 h of fraction treatment was studied. Cell cycle analysis by flow cytometry showed that nuclei in apoptotic cells decreased the DNA stain ability with PI, indicated by the appearance of a sub-G1 peak on the DNA histogram (Figure 4A). This result further converted into numeral histogram, shows the flow cytometry analysis on HL-60 cells stained with PI for 24h (Figure 4B). The apparition of sub-G1 peak was observed in 20% of the fraction II treated cells, which is 4 fold more than untreated control cells.
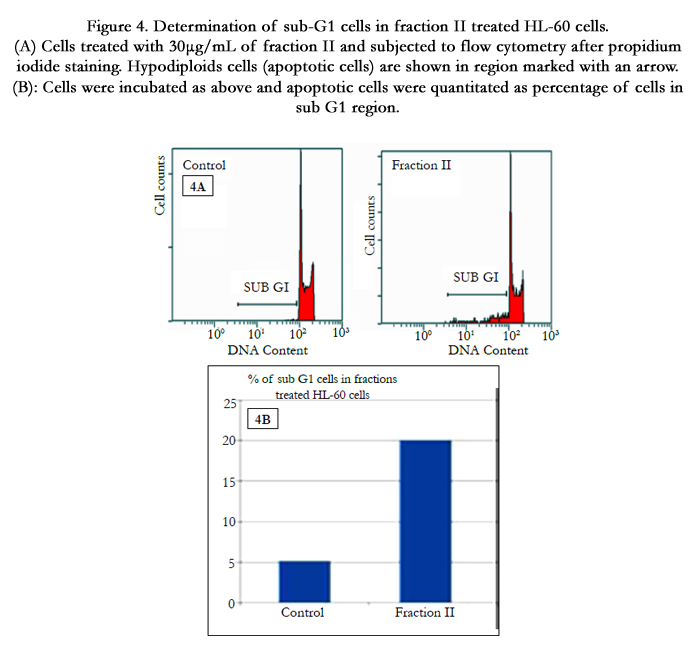
Figure 4. Determination of sub-G1 cells in fraction II treated HL-60 cells. (A) Cells treated with 30μg/mL of fraction II and subjected to flow cytometry after propidium iodide staining. Hypodiploids cells (apoptotic cells) are shown in region marked with an arrow. (B): Cells were incubated as above and apoptotic cells were quantitated as percentage of cells in sub G1 region.
Programmed cell death is another phenomenon that could explain inhibition in cell proliferation by analysis of DNA fragmentation, the latest stage of apoptosis. After 24h treatment, result showed that fraction II induced DNA fragmentation in HL-60 cells (Figure 5).
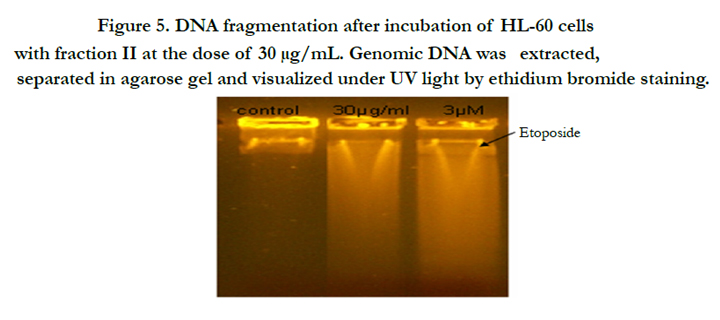

Figure 5. DNA fragmentation after incubation of HL-60 cells with fraction II at the dose of 30 μg/mL. Genomic DNA was extracted, separated in agarose gel and visualized under UV light by ethidium bromide staining.
In the light of the results of QNFM, cell cycle and DNA fragmentation, fraction II showed most potent activity and induces apoptotic cell death on HL-60 cells. On that background, chromatographic analysis was carried out only for fraction II.
Thin layer chromatographic analysis (Si-gel, CH2Cl2/MeOH, 20/1) of fraction II showed the presence of two major constituents. Then an attempt was directed to check the purity of fraction-II using HPLC-UV but, unfortunately, this method was unsuitable due to the weak absorbance of the compounds by UV. Therefore, the HPLC-ELSD technique was adopted. The ELSD chromatograms of fraction II showed 2 peaks, one low intense peak at 16 min and another one more intense at 16.5 min which confirms the presence of two compounds (Figure 6).
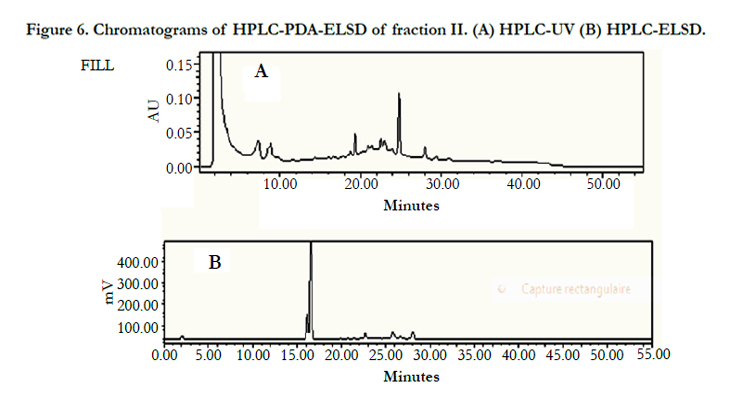
Figure 6. Chromatograms of HPLC-PDA-ELSD of fraction II. (A) HPLC-UV (B) HPLC-ELSD.
After purification by preparative HPLC, two compounds were analysed by GC-MS (Figure 7) and the results indicated the presence of three compounds, two known compounds are pentacyclic triterpenoids: oleanolic and ursolic acids and for identification of other compound need more investigation. The structural elucidation of ursolic acid (UA) and oleanolic (OA) isomers were carried out by comparison of experimental values from GC-MS with previously reported values [11].
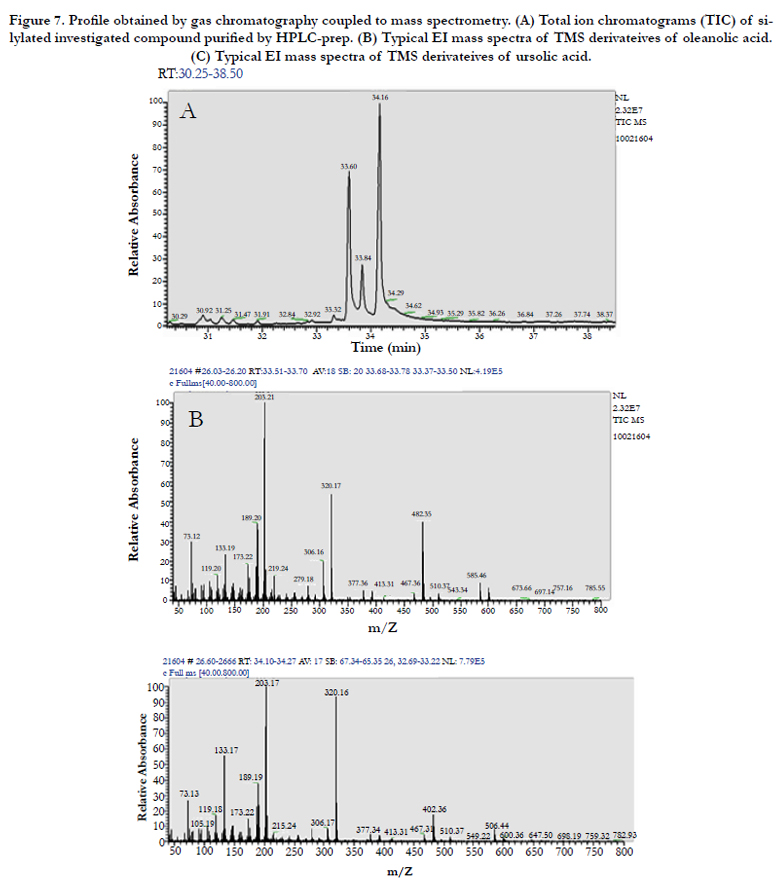
Figure 7. Profile obtained by gas chromatography coupled to mass spectrometry. (A) Total ion chromatograms (TIC) of silylated investigated compound purified by HPLC-prep. (B) Typical EI mass spectra of TMS derivateives of oleanolic acid. (C) Typical EI mass spectra of TMS derivateives of ursolic acid.
Discussion
Present study demonstrate that C. parqui leaves possess anticancer activity on HL-60 cell line. Chromatographic separation of the said extract was given eight fractions, out of which fraction II showed most potent efficacy in this concern. Phytochemical analysis of this fraction indicated that the actives compounds are oleanolic acid (OA) and ursolic acid (UA). These two compounds are pentacyclic triterpenoids having a similar chemical structure and are the major components of many medicinal plants and herbs widely distributed all over the world and these components have attracted a lot of attention because of their various biological activity such as anti-inflammatory, antioxidant, hepatoprotective, cytotoxic, antitumor and antiangiogenis [13-18]. So the proapoptotic effect of fraction II is not very surprising event because the presence of these antitumor compounds [19] but it’s mechanism of actions needs to more investigation. In the present study, it was found that 30μg/mLof fraction II inhibit cell proliferation and induced apoptotic cell death and the death is characterized by formation of apoptotic bodies, DNA fragmentation and cell cycle arrest in subG1 phase. To understand the pathway of apoptotic cell death, results were compared with the literature and two major pathways of apoptotic cell death program have been identified, namely receptor-mediated (extrinsic) and chemical-induced mitochondrial (intrinsic) apoptosis [20]. Zhang, et al. [21] reported that OA induced apoptotic cell death on HL-60 cell lines and the apoptotic death is characterized by formation of apoptotic bodies, DNA fragmentation, increased of the number of cell in the sub G1 phase, activation of caspase 3 and 9 and cleavage of poly (ADP ribose) polymerase and Takashi et al. [22] reported that OA inhibit the DNA topoisomerase, so the pathway of apoptotic death is extrinsic. Concerning the apoptotic effect of UA on HL-60, Beak et al. demonstrated that UA induced genomic DNA fragmentation, a hallmark of apoptosis, indicating that the mechanism by which UA induced cell death was through apoptosis [23]. They also demonstrated that UA induce the intracellular ca2+ release which may be involved in UA induced apoptosis in HL-60 cells [23]. So there is not much work regarding the mechanism of action of UA in the induction of apoptosis in HL-60 cells though several studies on other cell line such as gastric cancer cell line BGC-803, HT-29 , HepG2 human hepatoblastoma cell, human breast cancer cell line MDA-MB-231 [24-28].
Taken together the results, it may be suggest that the fraction II enriched with OA and UA induced the death via extrinsic pathway due to the dominance of the OA in these fraction as shown in the profile of GC-MS.
It is known that the mixture of OA and UA has a higher activity than individual compound but the compared with the literature, 30μg/mL of fraction II is more higher than OA and UA separately because EC50 of OA was 8.1μM and UA was 5.0μM [24]. This may be due to the presence of the other compound at low concentration but it may have antagonist activity to the OA and UA at this lower dose. Identification of the third compound needs further investigation.
One of the major side effects in the effective treatment of human malignancies is the acquisition of broad anti-cancer drug resistance by tumor cells. This phenomenon has been termed ‘‘multidrug resistance (MDR)’’. Therefore, development of new modulator for inhibition of drug resistance is required [29, 30]. So, to find out an effective drug that removes drug resistance from tumors, Shan Jian [31] demonstrated that OA and UA displayed significant inhibitory effects on HL-60 and HL60/ADR cells. Their cell proliferation inhibiting curves on parents cells and MDR cells of acute and chronic myelocytic leukemia were basically the same, the multiple of drug resistance being 1, suggesting that UA and OA are not the substrate of the drug pump proteins [31]. As a result they could not pumped out of the cells when entering and thus they were capable of displaying their implicit effect.
Furthermore, acute myeloid leukemia (AML) is characterized by a block in differentiation that leads to the accumulation of immature cells. So, the induction of differentiation is becoming the ideal theurapeutic strategy in a substrat of AML [32]. Alongside, UA was found to be an effective inducer of monocytic differentiation of HL-60 cells [33]. Taking together the results in the present study as well as previous findings, it may be assumed that OA and UA are important agents in the treatment of AML leukemia.
Conclusion
Finally, it was concluded that this is the first report about the existence of biologically active phytomolecules namely OA and UA in C. parqui leaves. These two phytomolecules exert anticancer activities on HL-60 cell line by two ways; one is apoptosis induction and other is inhibition in cell cycle. These two phytomolecules may be used as chemotherapy for certain cancer in future after extensive work in this line.
Acknowledgement
We are gratefully acknowledged to DST, Govt. of India, Science and Technology Division, Tunisia Govt. and Ministry of Education and Science of Spain for financial support.
References
- Zhao-you T (2012) Fighting against cancer by integrative medicine. Chin J Integr Med 18:323-324.
- Gottesman MM, Fojo T, Bates SE (2002) Multidrug resistance in cancer: role of ATP-dependent transporters. Nat Rev Cancer 2(1): 48-58.
- Choi CH (2005) ABC transporters as multidrug resistance mechanisms and the development of chemosensitizers for their reversal. Cancer Cell Int 5:30- 42.
- Baliga MS, Kurian PJ (2012) Ixora coccinea Linn.: Traditional uses, phytochemistry and pharmacology. Chin J Integr Med 18(1): 72-79.
- Quiroga EN, Sampietro AR, Vattuone MA (2001) Screening antifungal activities of selected medicinal plants. J Ethnopharmacol 74(1): 89-96.
- Fehmida TB, Akbar AA, Viqar UA (2001) Two new spirostonal glycosides from Cestrum parqui. Helvetica Chimica Acta 84: 3350-3356.
- D’Abrosca B, DellaGreca M, Fiorentino A, Monaco P, Oriano P, et al. (2004) Structure elucidation and phytotoxicity of C13 nor-isoprenoids from Cestrum parqui. Phytochem 65(4): 497-505.
- D’Abrosca B, DellaGreca M, Fiorentino A, Monaco P, Natale A, et al. (2005) Structural characterization of phytotoxic terpenoids from Cestrum parqui. Phytochem 66(22): 2681-2688.
- Rubio S, Quintana J, López M, Eiroa JL, Triana J, et al. (2006). Phenylbenzopyrones structure-activity studies identify betuletol derivatives as potencial antitumoral agents. Europe J Pharmacol 548(1-3): 9-20.
- Lee IS, Nishikawa A, Furukawa F, Kasahara K, Kim SU (1999) Effects of Selaginella tamariscina on in vitro tumor cell growth, p53 expression, G1 arrest and in vivo gastric cell proliferation. Cancer Lett 144(1): 93-99.
- Razborsek MI, Voncina DB, Dolecek V, Voncina E (2008) Determination of Oleanolic, Betulinic and Ursolic acid in Lamiaceae and mass spectral fragmentation of their trimethylsilylated derivatives. Chromatogram 67:433-440.
- Suffness M, Pezzuto JM (1990) Assays related to cancer drug discovery. In:Hostettmann K. (Ed.), Methods in Plant Biochemistry: Assays for Bioactivity. Academic Press, London. 6: 71–133.
- Banno N, Akihisa T, Tokuda H, Yasukawa K, Higashihara H, et al. (2004) Triterpene acids from the leaves of Perilla frutescens and their anti-inflammatory and antitumor-promoting effects, Biosci Biotechnol Biochem 68(1):85-90.
- Zdenka O, Katarına K, Darina S (2006) Protective effects of ursolic acid and oleanolic acid in leukemic cells. Mutation Res 600(1-2): 131–137.
- Saraswat B, Visen PKS, Dayal R, Agarwal DP, Patnaik GK (1996) Protective action of ursolic acid against chemical induced hepatotoxicity in rats. Indian J Pharmacol 28: 232–239.
- Min BS, Jung HJ, Lee JS, Kim YH, Bok SH, et al. (1999) Inhibitory effect of triterpenes from Crataegus pinatifida on HIV-I protease. Planta Med 65(4):374–375.
- Senthil S, Chandramohan G, Pugalendi KV (2007) Isomers (oleanolic and ursolic acids) differ in their protective effect against isoproterenolinduced myocardial ischemia in rats. Int J Cardiol 119(1): 131–33.
- Raphael TJ, Kuttan G (2003) Effect of naturally occurring triterpenoids glycyrrhizic acid, ursolic acid, oleanolic acid and nomilin on the immune system. Phytomed 10(6-7): 483–489.
- Tokuda H, Ohigashi H, Koshimizu K, Ito Y (1986) Inhibitory effects of ursolic acid and oleanolic acid on skin tumor promotion by 12-tetradecanoylphorbol- 13-acetate. Cancer Lett 33(3): 279–285.
- Xiaohua G, Dorrah D, Hao J, Yongbo L, Scott AD, et al. (2007) Synthetic triterpenoids inhibit growth and induce apoptosis in human glioblastoma and neuroblastoma cells through inhibition of prosurvival Akt, NF-jB and Notch1 signaling. J Neurooncol 84(2): 147–157.
- Zhang P, Li H, Chen D, Ni J, Kang Y et al. (2007) Oleanolic acid induces apoptosis in human leukemia cells through caspase activation and poly (ADP-ribose) polymerase cleavage. Acta Biochimica et Biophysica Sinica 39(10): 803–809.
- Takashi K, Hiroyuki A, Keiichi T, Aranya M, Jiradej M, et al. (2011) 3-O-(E)-p-Coumaroyl tormentic acid from Eriobotrya japonica leaves induces caspase-dependent apoptotic cell death in human leukemia cell line. Chem Pharm Bull 59(3): 378-381.
- Baek JH, Lee YS, Kang CM, Kim JA, Kwon KS et al (1997) Intracellular Ca2+ release mediates ursolic acid-induced apoptosis in human leukemic HL-60 cells. Int J Cancer 73(5): 725-728.
- Xuemei W, Fan Z, Ling Y, Ying M, Hai L, et al. (2011) Ursolic acid inhibits proliferation and induces apoptosis of cancer cells in vitro and in vivo. J Biomed Biotechnol 1-8.
- Jian-zhen S, Yan-yan X, Shu Z, Qi D, Su-zhan Z (2009) Ursolic acid inhibits proliferation and induces apoptosis of HT-29 colon cancer cells by inhibiting the EGFR/MAPK pathway. J Zhejiang Univ Sci B 10(9): 668-674.
- Kim DK, Baek JH, Kang CM, Yoo MA, Sung JW et al. (2000) Apoptotic activity of ursolic acid may correlate with the inhibition of initiation of DNA replication. Int J Cancer 87(5): 629–636.
- Kim KH, Seo HS, Choi HS, Choi I, Shin YC et al. (2011) Induction of apoptotic cell death by ursolic acid through mitochondrial death pathway and extrinsic death receptor pathway in MDA-MB-231 cells. Arch Pharm Res 34(8): 1363-1372.
- Manu KA, Kuttan G (2008) Ursolic acid induces apoptosis by activating p53 and caspase-3 gene expressions and suppressing NF-kappa B mediated activation of bcl-2 in B16F-10 melanoma cells. Int Immunopharmacol 8(7):974-981.
- Gottesman MM, Fojo T, Bates SE (2002) Multidrug resistance in cancer: role of ATP-dependent transporters. Nat Rev Cancer 2(1): 48-58.
- Choi CH (2005) ABC transporters as multidrug resistance mechanisms and the development of chemosensitizers for their reversal. Cancer Cell Int 5:30-42.
- Jian-zhen S, Yan-yan X, Shu-qin R, Mei S (2011) Proliferation-Inhibiting and Apoptosis-Inducing Effects of Ursolic Acid and Oleanolic Acid on Multi- Drug Resistance Cancer Cells in Vitro. Chin J Integr Med 17(8): 607-611.
- Nowak D, Stewart D, Koeffler HP (2009) Differentiation therapy of leukemia: 3 decades of development. Blood 113(16): 3655–3665.
- Ting Z, Yun-Mian H, Jin-Song W, Jing S, Ying-Ying X, et al. (2011) Ursolic acid induces HL-60 monocytic differentiation and upregulates C/EBPb expression by ERK pathway activation. Anti Cancer Drugs 22(2): 158–165.





