A Case of an Unusually Aggressive Cutaneous Anaplastic Large T-Cell Lymphoma in a Young Patient
S Elloudi1*, H Elmahi1, H Baybay1, S Gallouj1, M Rimani2, FZ Mernissi1
1 Department of Dermatology and Venereology, University Hospital Hassan II Fez, Morocco.
2 Centre pathologyHassane, Rabat, Morocco.
*Corresponding Author
S. Elloudi,
Department of Dermatology and Venereology , University Hospital Hassan II Fez, Morocco.
Tel: 212- 0667410849
E-mail: saraelloudi@gmail.com
Received: September 09, 2016; Accepted: October 17, 2016; Published: October 19, 2016
Citation: S Elloudi, H Elmahi, H Baybay, S Gallouj, M Rimani, FZ Mernissi (2016) A Case of an Unusually Aggressive Cutaneous Anaplastic Large T-Cell Lymphoma in a Young Patient. Int J Cancer Stud Res. 5(4), 111-114. doi: dx.doi.org/10.19070/2167-9118-1600021
Copyright: S Elloudi© 2016. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
Abstract
Primary Cutaneous Anaplastic Large-cell Lymphoma (PC-ALCL) is the second most common cutaneous T-cell Lymphoma. PCALCL is composed of large cells with an anaplastic, pleomorphic, or immunoblastic cytomorphology, and expression of the CD30 antigen by the majority (>75%) of tumor cells .Wehere by report a case of unusually aggressive PC-ALCL in a young adult.
2.Introduction
3.Case Report
4.Discussion
5.Conclusion
6.References
Keywords
Primary Cutaneous Anaplastic Large Cell Lymphoma; Anaplastic Lymphoma kinase; Aggressive, CD30.
Introduction
Primary Cutaneous Anaplastic large-Cell Lymphoma (PC-ALCL) is the second most common cutaneous T-cell Lymphoma [1]. (PC-ALCL) is part of the spectrum of CD30+ lymphoproliferative diseases of the skin including lymphomatoid papulosis. PCALCL is composed of large cells with an anaplastic, pleomorphic, or immunoblastic cytomorphology, and expression of the CD30 antigen by the majority (>75%) of tumorcells [2]. PC-ALCL remains confined to the skin with rare dissemination beyond local lymphnodes inmost cases [2]. PC-ALCL must be distinguished from secondary cutaneous lesions of systemic ALCL since the latter runs a poor clinical course [3]. PC-ALCL mainly affects older patients in the sixth decade. We here by report a case of unusually aggressive PC-ALCL in a young adult.
Case Report
A 34-year-old man with history of chronic smoking, admitted to our departement for pigmented patchs on the trunk and scalp lasting for 3 years (Figure 1). The patient reported spontaneous regression in some lesions (Figure 2). The large lesion is localized in the chest (Figure) began as a small reddened patch and after 3 month became a 30-cm, then 50cm raised, dark, ulcerative circular mass lesion (Figure 3). Lymphadenopathy was appreciated on physical examination. Histological and immunohistochemical study confirms primary cutaneous lymphoproliferation T CD30 (+) consistent with an anaplastic lymphoma cutaneous CD30 (+) Expressed by 75% of atypical cells (Figure 4, 5). ALK was negative. Tumor extension assessment had not objectified extra cutaneous reached. The histological study of surgical resection of axillary lymphadenopathy was inflammatory. The patient has received treatment by methotrexate, then CHOEP chemotherapy, without improvement. The patient died of septic shock.
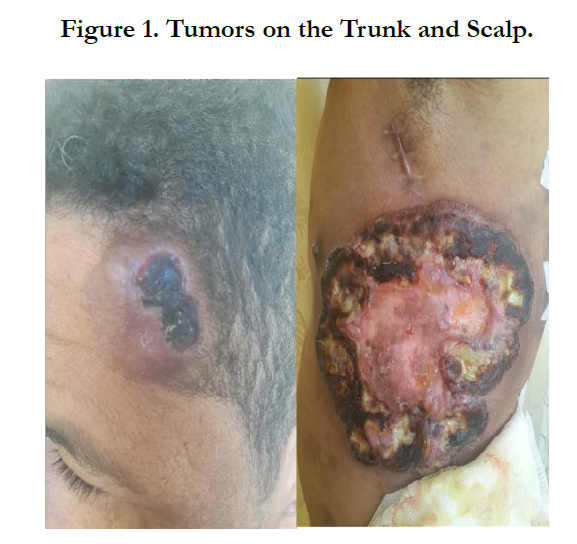
Figure 1. Tumors on the Trunk and Scalp.
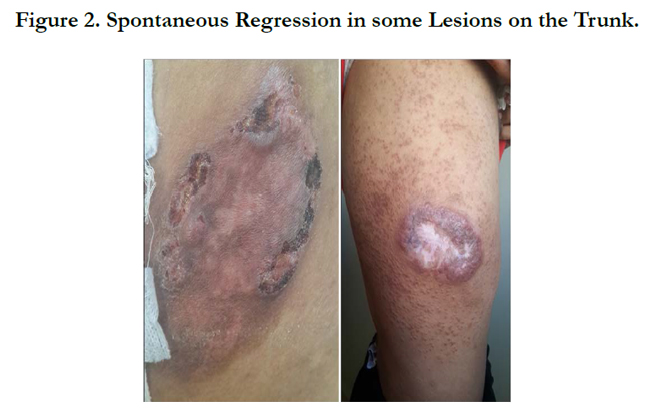
Figure 2. Spontaneous Regression in some Lesions on the Trunk.
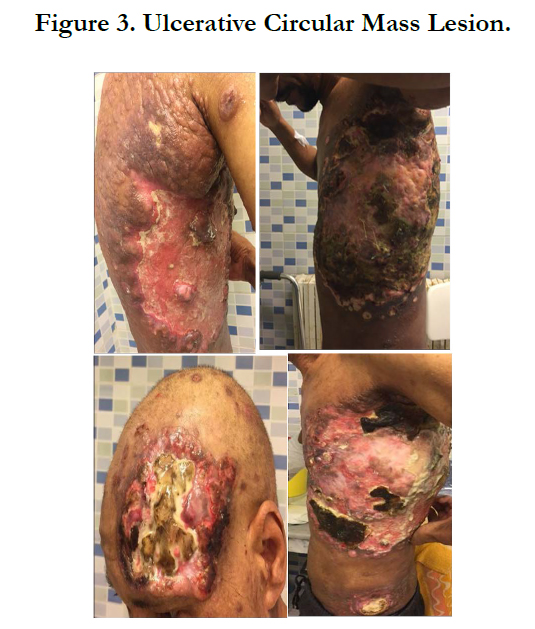
Figure 3. Ulcerative Circular Mass Lesion.
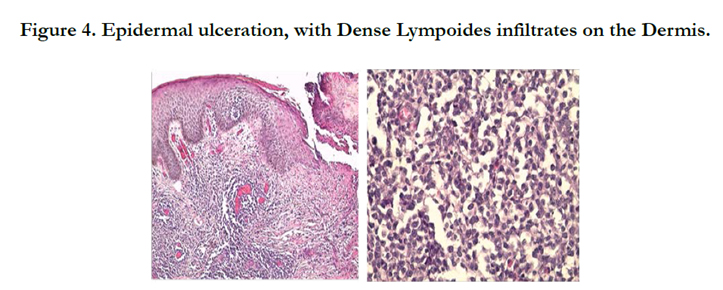
Figure 4. Epidermal ulceration, with Dense Lympoides infiltrates on the Dermis.
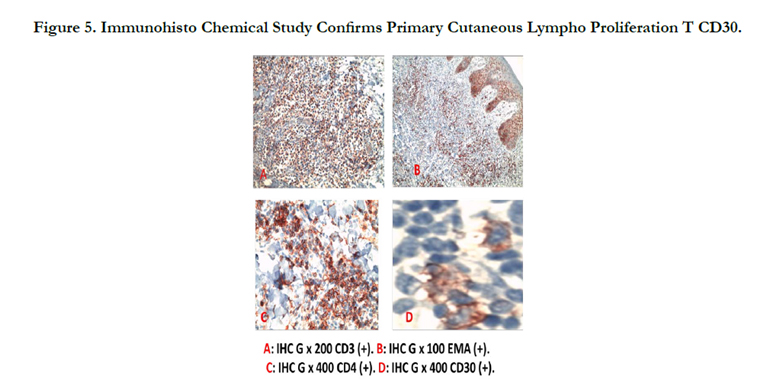
Figure 5. Immunohisto Chemical Study Confirms Primary Cutaneous Lympho Proliferation T CD30.
Discussion
PCALCL CD30-positive is the second most common form of cutaneous T-cell Lymphoma (CTCL) with an incidence of 0.1 to 0.2 patients per 100,000 [1]. It is mainly affects people in their sixth decade with a male to female ratio of 2 to 3/1 [1].
The criteria for the diagnosis of PCALCL include: more than 75% infiltration of CD30+ large anaplastic cells in skinbiopsy, no clinical history of lymphomatoid papulosis, mycosis fungoides or other cutaneous lymphomas, and no extra cutaneous localization after extensive investigations at presentation [4]. Our patient fulfilled all the criteria.
Clinically, it is solitary asymptomatic reddish to violaceous nodule, plaque or tumor over the head, trunk and extremities. The lesion is often slow growing with an indolent clinical course and may present for a long time before being diagnosed [5], and may regress spontaneously in 30% of cases [6]. However clinical presentation can be impressive with the rapid appearance of a single purplish red tumor with necrotic evolution. In our case, evolution were insidious for 3 years with the notion of spontaneous regression of lesions, then becoming rapidly progressive with installation of more extensive ulcerated and necrotic tumors.
Generalized or multifocal lesions are rare and seen in about 20% of the patients [2], they are not controlled by treatment with a risk of secondary progression extracutaneous high relative to the localized form (17% versus 8%), and a higher mortality rate (12% versus 3%) [7].
PC-ALCL is characterized histologically by dense cohesive sheets of large atypical cells, known as “hallmark cells”, comprising more than 75% of the cellular infiltrate. The cells have abundant cytoplasm with reniform-shaped hyperchromatic nuclei and prominent eosinophilic nucleoli with frequent mitoses [8], similar histological findings were observed in our case.
Nuclear matrix protein (NMP)-ALK is a nuclear fusion protein, whichis not expressed in normal lymphocytes. ALK expression is common in systemic ALCL and rare in PC-ALCL, its expression in skin is a warning to look for systemic disease [3] specially nodal involvement.
In our case, the ALK was negative, and the histological study of surgical resection of axillary lymphadenopathy was inflammatory confirming the primary cutaneous character of lymphoproliferation. Our patient is classified as stage T3bN0M0 based on the TNM classification system for primary cutaneous lymphomas other than MF/SS of the ISCL/EORTC [9].
Extracutaneous dissemination occurs in approximately 10% of patients with localized disease and mainly involves the regional lymphnodes, thus long-term follow-up is required in all patients with PC-ALCL.
The mechanisms that are involved in the development of anaplastic large cell lymphoma are unknown [10]. In most patients, the initial step that involves activation and clonal expansion of CD30+ T cells is controlled effectively by the host immune response [10].
Further progression occurs only when the tumor cells acquire a growth advantage, either by additional chromosomal alterations or when the host immune response becomes deficient [10]. However, there maybe spontaneous regression of the lesion if the host immune responseis intact [10].
PC-ALCL can lend confusion with transformed mycosis fungoides or with a lymphomatoid papulosis, hence the interest of a clinicopathological confrontation.
Treatment of PC-ALCL should be decided according to the size, number and extent of the tumor.
Primary C-ALCL is an indolent disease, therefore treatment measures should focus on noninvasive strategies. Localized radiation therapy or surgical excision for solitary or localized lesionsis the preferred treatment for C-ALCL [11]. Systemic therapy is indicated for patients refractory to local therapy, multifocal disease like our patient, and/or extracutaneous spread of disease. A well-tolerated option include slow-dose methotrexate [12, 13], but unfortunately it was not effective in our case. Alternative therapies such as oral bexarotene, Interferon‑alpha (IFN), thalidomide, or doxorubicin may be used for treating PC-ALCL [1, 14, 15].
Combination chemotherapy is not indicated in the first intension, but In view of an aggressive clinical course, and based on the available research [11-13], our patient was treated with chemotherapy type CHOEP. Unfortunately, this tumor has a tendency to recur, even after long disease-free intervals. According to the literature review, multi agent systemic chemotherapy does not prevent future relapses [7].
Newer classes of drugs such as brentuximab vedotin (SGN35) [16, 17], histone deacetylase (HDAC) inhibitors are now available as treatment alternatives. Recently Romidepsin was approved for treatment of cutaneous ALCL for patients that had received systemic chemotherapy at least once [18].
In our patient, the refractory and debilitating multifocal shape was enough to indicate multiagent systemic chemotherapy. Moreover, the vital prognosis can be put into play by the iatrogenic immunosuppression.
Conclusion
Although they are exceptional, multifocal, recurrent and refractory forms seem very debilitating, having a fatal prognosis, and pose a problem of management.
References
- Kempf W, Pfaltz K, Vermeer MH, Cozzio A, Bagot M, et al., (2011) EORTC, ISCL, and USCLC consensus recommendations for the treatment of primarycutaneous CD30-positive lymphoproliferativedisorders: lymphomatoidpapulosis and primary cutaneous anaplastic large-celllymphoma. Blood. 118: 4024-4035.
- Willemze R, Jaffe ES, Burg G, Cerroni L, Berti E, et al., (2005) WHOEORTC classification for cutaneous lymphomas. Blood. 105: 3768-3785.
- Diamantidis MD, Myrou AD (2011) Perils and pitfallsregardingdifferentialdiagnosis and treatment of primary cutaneous anaplastic large-celllymphoma. Sci World J. 11: 1048-55.
- Gould J, Eppes R, Gilliam A, et al., (2000) Solitary primary cutaneous CD30+ large cell lymphoma of natural killer cell phenotype bearing the t(2;5)(p23;q35) translocation and presenting in a child. Am J Dermatopathol.22(5): 422-428.
- Sridevi HB, Shanthala PR, Raghuveer CV, Prabhu AK, Akbar JK, et al., (2015) A rare case of ALK negativeCD30+ primary cutaneous anaplastic large cell lymphoma in a young adult. J Can Res Ther. 11(3): 656.
- Vergier B, Beylot-Barry M, Pulford K, Michel P, Bosq J, et al., (1998) Statistical evaluation of diagnostic and prognostic features of CD30+ cutaneouslymphoprliferative disorders : aclinicopathologic study of 65 cases. Am J Surg Pathol. 22: 1192-202.
- Bekkenk MW, Geelen F, Heule F, Geerts ML, Meijer CJ, et al., (2000) Primary and secondary ctaneous CD30+ lymphoproliferative disorders : a report from the Dutch cutaneous lymphoma Group on the long-termfollowup data of 219 pateints, end guidelines for diagnosis and treatment. Blood. 95(12): 3653-61.
- Pauline GM, Chao-Lo, King-Ismael D, Lopez RA (2008) Primary cutaneous CD30+anaplastic large cell lymphoma: Report of a rare case. J Dermatol Case Rep. 2(3): 31-34.
- Kim Y, Willemze R, Pimpinelli N, Olsen EA, Ranki A, et al., (2007) TNM classification system for primary cutaneous lymphomas other than mycosis fungoides and Sezary syndrome: a proposal of the International Society for Cutaneous Lymphomas (ISCL) and the Cutaneous Lymphoma Task Force of the European Organization of Research and Treatment of Cancer (EORTC).Blood. 110: 479-484.
- Willemze R, Meijer C (2003) Primary cutaneous CD30-positive lymphoproliferative disorders. Hematol Oncol Clin North Am. 17(6): 1319-1332.
- MA Fung, MJ Murphy, DM Hoss, JM Grant- Kels (2002) Practical evaluation and management of cutaneous lymphoma. J Am Acad Dermatol. 46(3): 325–357.
- Kadin ME, Carpenter C (2003) Systemic and primary cutaneous anaplastic large cell lymphomas. Semin Hematol. 40(3): 244-256.
- Vonderheid EC, Sajjadian A, Kadin ME (1996) Methotrexate is effective therapy for lymphomatoid papulosis and other primary cutaneous CD30- positive lymphoproliferative disorders. J Am Acad Dermatol. 34(3): 470-481.
- French LE, Shapiro M, Junkins JM, Vittorio CC, Rook AH (2001) Regression of multifocal, skin-restricted, CD30 + large T-cell lymphoma with interfer on alfa and bexarotène therapy. J Am Acad dermatol. 45(6): 914-918.
- Olivera A, Alves R (2011) Primary cutaneous CD30+ anaplastic large cell lymphoma-reprt of a case treated by bexarotène. Leuk Res. 35(11): 190- 1902.
- Younes A, Barlett NL, Leonard JP, Kennedy DA, Lynch CM, et al., (2010) Brentuximab vedotin (SGN-35) for relapsed CD30+ lymphoma. Neng J Med. 363(19: 1812-1821.
- Querfeld C, Khan I, Mahon B, Nelson BP, Rosen ST et al., (2010) Primary cutaneous and systemic anaplastic large cell lymphoma: clinicopathologic aspects and therapeutic options. Oncology. 24(7): 574–587.
- Vose JM (2010) Understanding evaluation and management of primary cutaneous and systemic ALCL. Oncology. 24(7): 594.





