Acrylonitrile Induced Alterations in Mitochondrial Biogenesis and Bioenergetics in Cultured Rat Astrocytes
Wang Z, Zhou S*, Pu X, Klaunig JE*
Department of Environmental Health, Indiana University School of Public Health, Bloomington, USA.
*Corresponding Author
Shaoyu Zhou Ph.D, and James E. Klaunig Ph.D,
Department of Environmental Health,
Indiana University, 1025 East 7th street,
Bloomington, IN 47405, USA.
Tel: 812-856-4936
Fax: 812-856-2445
E-mail: zhous@indiana.edu and jklauni@indiana.edu
Article Type: Research Article
Received: May 10, 2015; Accepted: June 22, 2015; Published: June 25, 2015.
Citation: Wang Z, Zhou S, Pu X, Klaunig JE (2015) Acrylonitrile Induced Alterations in Mitochondrial Biogenesis and Bioenergetics in Cultured Rat Astrocytes. Int J Cancer Stud Res. 4(3), 75-81. doi: dx.doi.org/10.19070/2167-9118-1500011
Copyright: Zhou S and Klaunig JE © 2015. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
Abstract
Acrylonitrile (ACN) induces brain tumors in rats following chronic exposure. The induction of oxidative stress may be involved in the mechanism of ACN induced carcinogenesis although the exact mechanism remains to be elucidated. The present study was designed to examine whether ACN induces oxidative mitochondrial DNA (mtDNA) damage and alterations in mitochondrial bioenergetics and biogenesis in cultured rat astrocytes. Cells were treated with ACN for 24 hr, 48 hr, or 14 wk, respectively, to examine the acute and chronic effect of ACN. Oxidative mtDNA damage was detected via measuring 8-hydroxydeoxyguanosine (8-OHdG) with high performance liquid chromatography. MtDNA content was quantitated with real-time PCR, while mitochondrial membrane potential and apoptotic cell death were examined by flow cytometry. Astrocytes treated with ACN for 24 hr resulted in a significant increase in oxidative mtDNA damage, which was persistent after 14 wk. Similarly, ACN treatment of astrocytes for 24 hr resulted in a dose-dependent increase in mtDNA content, which remained significantly increased after 14 wk treatment. In addi tion, we showed that astrocytes treated with ACN for 14 week exhibited hyperpolarization in mitochondrial membrane potential, suggesting that chronic treatment causes a persistent change in mitochondrial bioenergetics. Furthermore, we demonstrated the cells from chronic ACN treatment exhibited resistance to apoptotic cell death induced by doxorubicin and cisplatin. Taken together, these data suggest that ACN treatment induces persistent oxidative mtDNA damage and sustained mitochondrial bioenergetic and biogenic changes, which may be involved in the mechanism of ACN induced carcinogenesis.
2.Introduction
3.Materials and Methods
3.1.Chemicals and cell culture
3.2.Isolation of mitochondrial DNA from cultured astrocytes
3.3.Analysis of 8OHdG
3.4.Determination of mitochondrial membrane potential
3.5.Quantitative Real-Time PCR
3.6.Statistical analysis
4.Results
4.1.ACN treatment increased oxidative mtDNA damage
4.2.ACN treatment resulted in persistent increase in mitochondrial DNA content
4.3.14 wk ACN treatment changed mitochondrial membrane potential
4.4.Chronic ACN treatment conferred resistance to cytotoxicity of anti-cancer agents
5.Discussion
6.Acknowledgement
7.References
Keywords
Acrylonitrile; Mitochondrial DNA; Oxidative Stress; Brain Tumor.
Introduction
Acrylonitrile (ACN) is widely used in the manufacture of acrylics, plastics and synthetic rubber. Early investigation suspected that acrylonitrile exposure might possess a risk of causing human brain tumor. For example, epidemiological investigation found that workers in the petrochemical industry and, specifically, in the production of PVC or acrylonitrile-based polymers had an increased risk of brain tumor [1]. More studies have found that no increased incidents of brain tumors occurred in workers exposed to ACN [7, 15]. In rodents, however, studies have demonstrated that chronic exposure to acrylonitrile caused dose-related increases in brain tumors in rats [2, 12, 24]. Yet, molecular mechanisms behind ACN elicited carcinogenesis in rodents remain largely unknown. Since there is potential human exposure especially among occupational workers, ACN remains an important environmental issue and studies of mechanism of ACN carcinogenesis are of significance.
Oxidative stress has been proposed to be involved in the mechanism of ACN-induced brain tumor in rodents. Our laboratory previously found that ACN treatment resulted in a dose-related increase in oxidative DNA damage in cultured astrocytes as well as in ACN exposed rats. Furthermore, we demonstrated that this ACN induced increase in oxidative DNA damage can be reduced/ prevented by antioxidant supplementation [22, 23]. These results support the involvement of the induction of oxidative stress and damage in the mode of action for acrylonitrile - induced astrocytomas in rodents.
The role for the mitochondria as a target of environmental agents has been widely implicated into the mechanism of chronic exposure to environmental toxicants. Mitochondria are important cellular source of reactive oxygen species (ROS), and on the other side, are susceptible to the attack of chemical induced oxidative stress, which may result in mitochondrial bioenergetics and mitochondrial genetic alterations that have been frequently observed in environmental agents-induced toxicities [19]. Studying effects of ACN on mitochondria thus may offer a new avenue for the understanding of the molecular mechanism of oxidative stress mediated ACN carcinogenesis. Indeed, the role of dysfunctional mitochondria in carcinogenesis has recently re-emerged because of a proposed regulatory role of mitochondria in apoptosis and ROS production. Somatic mutations and large deletions of mtDNA have been increasingly proposed to participate in carcinogenesis by affecting mitochondrial functions resulting in increase in ROS generation and dysregulation of apoptosis. One of the mechanisms causing mtDNA mutations may involve in oxidative DNA damage when not repaired properly. It has been well documented that ACN causes oxidative DNA damage in genomic DNA [22,23]. However, whether ACN treatment causes oxidative mtDNA damage has yet to be determined. It is believed that mtDNA is more susceptible to DNA damage and consequently acquires mutations at a higher rate than does nuclear DNA. Several possible factors may account for these differences in susceptibility including exposure to high levels of ROS produced during oxidative phosphorylation, lack of protective histones, and limited capacity for repair of DNA damage [20, 6, 3]. Thus, investigation of oxidative mtDNA damage may provide additional input into our understanding of the molecular mechanism of ACN carcinogenesis.
It is suggested that mitochondria may also be involved in carcinogenesis through alteration of biogenesis. A number of studies have demonstrated that alterations in mitochondrial contents are associated with different types of cancers including lung, head and neck cancers [14, 11, 4]. Although the mechanism of altered mitochondrial biogenesis in carcinogenesis is not described, it may be related to alteration in mitochondrial function that gives rise to increased mitochondrial ROS generation and altered regulation of mitochondrial apoptosis. Studies on genomic DNA have suggested ACN to be a nongenotoxic carcinogen; however, concrete conclusion has not been achieved. On the other hand, to date the effect of ACN on mitochondrial biogenesis and bioenergetics has not been examined.
In the present study, therefore, we determined ACN induced oxidative mtDNA damage and investigated ACN mediated alteration in mtDNA content in astrocytes. We also investigated whether cells chronically treated with ACN were resistant to apoptotic inducers, which is one of the mechanisms implicated in chemical carcinogenesis.
Materials and Methods
Acrylonitrile was obtained from INEOS Nitriles (League City, Texas). 8-hydroxy-2’-deoxyguanosine (8OHdG), 2-deoxyguanosine (2dG), NaI, deferoxamine mesylate, and nuclease P1 were purchased from Sigma-Aldrich (St. Louis, MO).
DI TNC1 rat astrocytes (ATCC, Rockville, MD) were cultured in DMEM/F12 medium supplemented with 4.5 g/L glucose, 10% FBS, penicillin (200 U/mL), streptomycin (200 μg/ml) and Amphotericin B (25 μg/mL), at 37°C with 5% CO2. Cells were plated at a density of 1×105 in 35 mm culture dishes. For long-term treatment, cells were treated with 0.25 mM ACN, and medium was changed every two to three days with the addition of fresh ACN. Cells were subcultured when they reached 75 to 100% confluency.
Cells (5 x 107) were harvested by trypsinization. Following trypsinization, cells were re-suspend in ice cold PBS and then centrifuged for 5 minutes at 600 g at 2–8°C. The pellets were suspended in ice cold isolation buffer and then homogenized with Dounce homogenizer. The homogenates were centrifuged for 10 min at 1,000 g at 4°C, and the supernatants were centrifuged for 10 min at 10, 000 g at 4°C. The resultant pellets were then subjected to digestion by Proteinase K at 50°C for 1 hr. And DNA was precipitated by NaI, and resultant DNA pellet is dissolved in dH2O.
DNA samples were processed according to standard protocol and analyzed for 8-OHdG by HPLC as described previously [23]. Analysis of 2-deoxyguanosine (2-dG) was detected at 290 nm (Waters 996 system; Waters, Milford, MA). 8-OHdG and 2-dG were quantitated from standards prepared in mobile phase immediately prior to sample analysis.
Mitochondrial membrane potential was determined by flow cytometry using the ΔΨm-sensitive fluorescent dye JC-1 (5,5'6,6'-tetrachloro- 1,1',3,3' tetraethylbenzimidazol carbocyanineiodide; Molecular Probes, Eugene, OR), analyzed in fluorescence detection channel 2 (FL-2), as described previously [9].
The mitochondrial DNA content was determined by amplification of mitochondrial gene ATP8 and D-loop with 7900HT Fast Real-time PCR Systems (Applied Biosystems). The real-time PCR reactions were performed in triplicate for each gene. DNA (5 ng) was used to amplify mitochondrial regions, whereas 5 ng of DNA was used to amplify β-actin as a control. Data analysis was performed using Microsoft EXCEL software. mtDNA/nDNA ratios were calculated by dividing the mtDNA signal for each gene by the β-actin signal and expressing the ratio as a percentage of the untreated control set at 100%.
Data were presented as mean ± SD of three individual experiments. Differences between treatment and control were evaluated using Students’ t-test. A p value of <0.05 was considered as significance.
Results
To determine whether ACN causes increased mtDNA damage, 8-OHdG levels in isolated mtDNA were analyzed using HPLC as described previously with modifications [23]. As shown in Figure 1, 24 hr treatment of ACN resulted in a dose-dependent increase in 8-OHdG level, indicating oxidative damage to mtDNA (Figure1A). Since cells have a line of defense mechanisms that may be upregulated in response to increased oxidative stress, continuous exposure to oxidative stress may increase the expression of antioxidant mechanism which prevents or reduces oxidative DNA damage. Therefore, we determined the effect of chronic ACN treatment on mtDNA damage. A concentration of 0.25 mM was selected for the long-term treatment since higher concentrations caused serious cytotoxicity. While a modest increase in oxidative DNA damage was observed at 24 hr treatment, ACN at 0.25 mM caused significant increase in 8-OHdG level in mitochondrial DNA following chronic treatment (14 wk), suggesting a persistent oxidative mtDNA damage (Figure 1B).
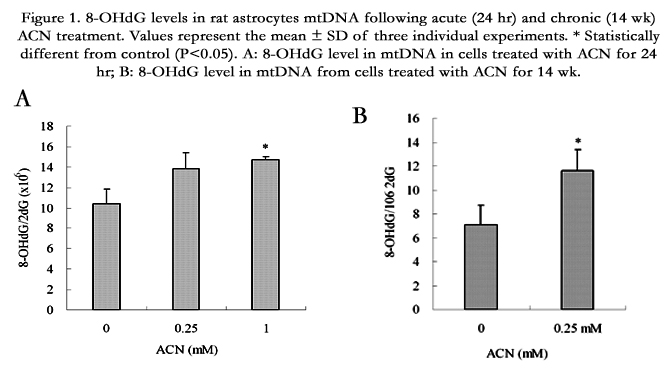
Figure 1. 8-OHdG levels in rat astrocytes mtDNA following acute (24 hr) and chronic (14 wk) ACN treatment. Values represent the mean ± SD of three individual experiments. * Statistically different from control (P<0.05).
A: 8-OHdG level in mtDNA in cells treated with ACN for 24 hr;
B: 8-OHdG level in mtDNA from cells treated with ACN for 14 wk.
To assess the effect of ACN on mtDNA content, cells were treated with ACN for 24 and 48 hr. Real-time PCR was performed on mitochondrial gene ATP8 and D-loop. It was found that 24 hr treatment induced a significantly dose-dependent increase in mtDNA content (Figure 2A). Furthermore, the increased mtDNA content was observed in cells treated with ACN for 14 wk, (Figure 2C) suggesting a persistent effect of ACN on mitochondrial biogenesis (Figure 2B).
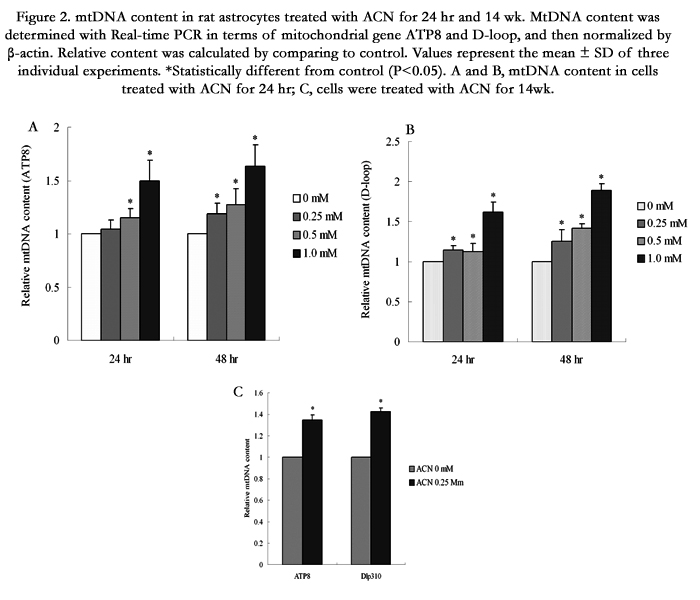
Figure 2. mtDNA content in rat astrocytes treated with ACN for 24 hr and 14 wk. MtDNA content was determined with Real-time PCR in terms of mitochondrial gene ATP8 and D-loop, and then normalized by β-actin. Relative content was calculated by comparing to control. Values represent the mean ± SD of three individual experiments. *Statistically different from control (P<0.05). A and B, mtDNA content in cells treated with ACN for 24 hr; C, cells were treated with ACN for 14 wk.
Both oxidative mtDNA damage and alteration in mtDNA contend may result in biological function change in mitochondria. To examine whether ACN can induce changes in mitochondrial function in ACN treated astrocytes, mitochondrial membrane potential was determined. The results showed that long-term treatment resulted in hyperpolarization (Figure 3), one of the common features shared by cancer cells [17].
Figure 3. Effect of ACN on mitochondrial membrane potential. Mitochondrial membrane potential was determined flow cytometry with fluorescent probe JC-1. Values represent the mean ± SD of three individual experiments. * Statistically different from control (p<0.05). A, a representative graph showing determination of membrane potential by flow cytometry; B, mitochondrial membrane potential in cells treated with ACN for 14 weeks.
To determine whether the mitochondrial hyperpolarization induced by chronic ACN treatment affects the regulation of apoptosis, we treated cells with doxorubicin (DOX), an antitumor drug known to induce apoptosis [16]. As shown in Figure 4, cells from long-term treatment with 0.25 mM ACN exhibited higher viability in response to DOX treatment compared to control cells (Figure 4). To gain more insights on the mechanism of resistance of cells, we treated cells that exposed to ACN for 14 wk with cisplatin, another antitumor agent that is known to induce apoptotic cell death. As shown in Figure 5, a significant resistance was associated with cells from long-term ACN treatment compared to control cells (Figure 5).
Figure 4. Cells treated with ACN for 14 weeks exhibited resistance to DOX-induced cytotoxicity. Values represent the mean ± SD of three individual experiments. * Statistically different from control (p<0.05).
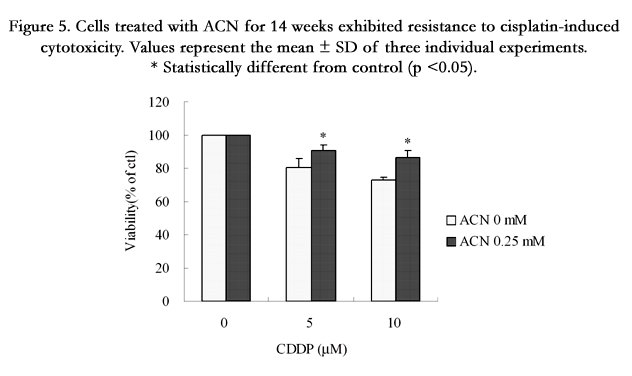
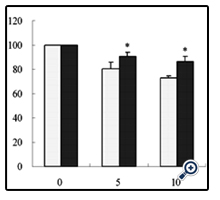
Figure 5. Cells treated with ACN for 14 weeks exhibited resistance to cisplatin-induced cytotoxicity. Values represent the mean ± SD of three individual experiments. * Statistically different from control (p <0.05).
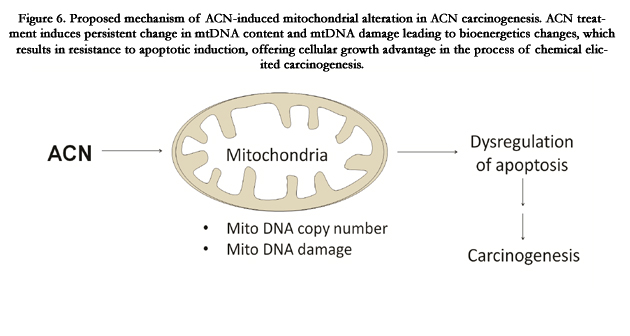
Figure 6. Proposed mechanism of ACN-induced mitochondrial alteration in ACN carcinogenesis. ACN treatment induces persistent change in mtDNA content and mtDNA damage leading to bioenergetics changes, which results in resistance to apoptotic induction, offering cellular growth advantage in the process of chemical elicited carcinogenesis.
Discussion
Acrylonitrile (ACN) is a well-established carcinogen in rodents that causes brain tumor in rats following chronic exposure [2, 12, 24]. It is proposed that oxidative stress induced by ACN may be involved in carcinogenesis although the molecular mechanism remains to be elucidated. Previous studies demonstrated ACN causes oxidative DNA damage both in vitro and in vivo [13, 22,23]. Since mitochondrial DNA is susceptible to oxidative DNA damage, therefore, the present study was designed to investigate whether ACN causes mtDNA damage and induces mitochondrial bioenergetic and biogenic changes in cultured rat astrocytes.
The results from the present study demonstrated that ACN caused increased 8-OHdG oxidative mtDNA damage (Figure 1A). More importantly, the oxidative mtDNA damage was not transient, but rather persistent over a period of 14 wk of ACN exposure (Figure 1B). Since oxidative stress may induce an array of cellular defense mechanism and DNA repair enzymes that protect DNA from oxidative damage, the persistent oxidative mtDNA damage suggests an insufficient induction of antioxidants and/or lack of efficient DNA damage repair mechanisms. A comprehensive investigation of antioxidants and repair enzymes may reveal more mechanistic insights into the persistence of oxidative mtDNA damage observed in astrocytes in response to ACN treatment. It has been reported that oxidative DNA damage, particularly 8-OHdG, is tumorigenic [26]. In this regards, increase in 8-OHdG level in mtDNA of astrocytes treated with ACN may consequently cause mitochondrial DNA mutations. There has been an increasing interest in role of mitochondrial mutations in carcinogenesis and tumorigenesis. Mutations in mtDNA are reported in almost all types of cancers examined so far [8, 5]. Although exact molecular mechanisms are complex, mutations in mtDNA have been demonstrated to actively contribute tumorigenesis and even metastasis [21, 28, 10]. Thus it is conceivable that ACN-induced oxidative mtDNA may lead to mtDNA mutations which may play a role in carcinogenesis. Further investigations, such as mtDNA sequencing, are needed to reveal more mechanistic insights into the ACN carcinogenesis. Another finding in the present study is the increase in mtDNA content in cells treated with ACN. The molecular mechanism that causes increase in mtDNA content remains unclear. It has been suggested to be related to oxidative stress and oxidative DNA damage which may activate replication and/or transcription of mtDNA [18, 25]. Interestingly, the increase in mtDNA content was found to be persistent over the 14 wk period that was correlated with the pattern of oxidative mtDNA (Figure 1 and 2), suggesting that oxidative mtDNA may causatively account for changes in mtDNA content. However, it should be noted that we were not able to distinguish whether the increase in mitochondrial DNA content was due to the increase in total number of mitochondria in a cell, or simply the increase in replication of mtDNA, thus more DNA copies in each mitochondrion. Nevertheless, we demonstrated a significant increase in mitochondrial DNA content caused by ACN. Alterations in mtDNA content have been documented in a number of tumors. Study has demonstrated that mitochondrial DNA quantity increases with histopathologic grade in premalignant and malignant head and neck lesions [14]. More recently, it was shown that mtDNA content was positively correlated with lung cancer risk [4]. However, it is unclear whether alteration in mtDNA content plays a role in carcinogenesis.
Despite the uncertain role of mtDNA content in carcinogenesis, the change in mtDNA may result in functional change of mitochondria. Since mtDNA encodes 13 important proteins located in mitochondrial electron transport chain (ETC), we proposed that increase in mtDNA content may result in alteration in expression of these important proteins, thus change mitochondrial biological function. One of the important characteristics of mitochondria is mitochondrial membrane potential which is closely related to function of ETC. In the present study, we examined the effect of ACN on mitochondrial membrane potential. Although acute treatment of cells with ACN caused a decrease in mitochondrial membrane potential (data not shown), the chronic treatment of ACN resulted in hyperpolarization (Figure 3), which confers selective advantage over long-term treatment of ACN. While exact mechanism has yet to be defined, as illustrated in Figure 6, we postulated that ACN treatment induces persistent change in mtDNA content and mtDNA damage that in turn leads to mitochondrial hyperpolarization. This alteration in mitochondrial bioenergetics would enhance cellular resistance to apoptotic induction, offering growth advantage in the process of chemical elicited carcinogenesis.
It has been demonstrated that mitochondrial membrane potential change is one of the common features of cancer cells [17]. A hyperpolarization of mitochondrial membrane potential can reduce the propensity of induction of mitochondrial permeability transition [29], a mitochondrial phenomenon that is frequently implicated into mitochondrial apoptosis. Indeed, dysregulation of apoptosis has been widely implicated into carcinogenesis and tumor progression, and cancer cells in general develop resistance to mitochondrial apoptosis [17]. Therefore, we determined response of cells from chronic ACN exposure to DOX and cisplatin, two antitumor drugs. We found that chronic ACN treatment conferred cells significant resistance to DOX and cisplatin induced cytotoxicity (Figure 4, 5). A number of reports show that DOX and cisplatin induce mitochondrial mediated apoptotic cell death [27, 16], and preserving mitochondrial function protects cells against cytotoxicity induced by DOX and cisplatin. The precise mechanism that chronically treated cells develop resistance to cytotoxicity induced by antitumor agents remains unknown. It may be related to the increase in mtDNA and hyperpolarization. MtDNA mutations may also affect mitochondrial protein expression resulting in bioenergetic change implicated into the mechanism of resistance to cytotoxicity of antitumor agents.
In summary, the present study demonstrated that ACN affected both mitochondrial biogenesis and bioenergetics in cultured rat astrocytes. ACN caused persistent oxidative mtDNA damage and increased mtDNA content. Chronic treatment of astrocytes with ACN resulted in a mitochondrial hyperpolarization. Furthermore, these chronic treated cells displayed resistance to antitumor agents induced cytotoxicity. All these alterations in mitochondria confer survival advantages that would play a significant role in the process of tumor formation and progression caused by ACN.
Acknowledgement
This study was supported in part by the Acrylonitrile Group Inc.
References
- Beall C, Delzell E, Rodu B, Sathiakumar N, Myers S (2001) Cancer and benign tumor incidence among employees in a polymers research complex. J Occup Environ Med 43(10): 914-924.
- Bigner DD, Bigner SH, Burger PC, Shelburne JD, Friedman HS (1986) Primary brain tumours in Fischer 344 rats chronically exposed to acrylonitrile in their drinking-water. Food Chem Toxicol 24(2): 129-137.
- Boiteux S, Radicella JP (1999) Base excision repair of 8-hydroxyguanine protects DNA from endogenous oxidative stress. Biochimie 81(1-2): 59-67.
- Bonner MR, Shen M, Liu CS, Divita M, He X et al. (2009) Mitochondrial DNA content and lung cancer risk in Xuan Wei, China. Lung Cancer 63(3):331-334.
- Chatterjee A, Mambo E, Sidransky D (2006) Mitochondrial DNA mutations in human cancer. Oncogene 25(34): 4663-4674.
- Chen CH, Cheng YC (1992) The role of cytoplasmic deoxycytidine kinase in the mitochondrial effects of the anti-human immunodeficiency virus compound, 2',3'-dideoxycytidine. J Biol Chem 267(5): 2856-2859.
- Cole P, Mandel JS, Collins JJ (2008) Acrylonitrile and cancer: a review of the epidemiology. Regul Toxicol Pharmacol 52(3): 342-351.
- Fliss MS, Usadel H, Caballero OL, Wu L, Buta MR, et al. (2000) Facile detection of mitochondrial DNA mutations in tumors and bodily fluids. Science 287(5460): 2017-2019.
- Heerdt BG, Houston MA, Augenlicht LH (2005) The intrinsic mitochondrial membrane potential of colonic carcinoma cells is linked to the probability of tumor progression. Cancer Res 65(21): 9861-9867.
- Ishikawa K, Takenaga K, Akimoto M, Koshikawa N, Yamaguchi A, et al. (2008) ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science 320(5876): 661-664.
- Jiang WW, Masayesva B, Zahurak M, Carvalho AL, Rosenbaum E, et al. (2005) Increased mitochondrial DNA content in saliva associated with head and neck cancer. Clin Cancer Res 11(7): 2486-2491.
- Johannsen FR, Levinskas GJ (2002) Chronic toxicity and oncogenic doseresponse effects of lifetime oral acrylonitrile exposure to Fischer 344 rats. Toxicol Lett 132(3): 221-247.
- Kamendulis LM, Jiang J, Xu Y, Klaunig JE (1999) Induction of oxidative stress and oxidative damage in rat glial cells by acrylonitrile. Carcinogenesis 20(8): 1555-1560.
- Kim MM, Clinger JD, Masayesva BG, Ha PK, Zahurak ML, et al. (2004) Mitochondrial DNA quantity increases with histopathologic grade in premalignant and malignant head and neck lesions. Clin Cancer Res 10(24):8512-8515.
- Klaunig JE (2008) Acrylamide carcinogenicity. J Agric Food Chem 56(15): 5984-5988.
- Konorev EA, Vanamala S, Kalyanaraman B (2008) Differences in doxorubicin- induced apoptotic signaling in adult and immature cardiomyocytes. Free Radic Biol Med 45(12): 1723-1728.
- Kroemer G, Pouyssegur J (2008) Tumor cell metabolism: cancer's Achilles' heel. Cancer Cell 13(6): 472-482.
- Lee HC, Lu CY, Fahn HJ, Wei YH (1998) Aging- and smoking-associated alteration in the relative content of mitochondrial DNA in human lung. FEBS Lett 441(2): 292-296.
- Meyer JN, Leung MC, Rooney JP, Sendoel A, Hengartner MO, et al. (2013) Mitochondria as a target of environmental toxicants. Toxicol Sci 134(1): 1-17.
- Moriya M, Ou C, Bodepudi V, Johnson F, Takeshita M, et al. (1991) Sitespecific mutagenesis using a gapped duplex vector: a study of translesion synthesis past 8-oxodeoxyguanosine in E. coli. Mutat Res 254(3): 281-288.
- Petros JA, Baumann AK, Ruiz-Pesini E, Amin MB, Sun CQ, et al. (2005) mtDNA mutations increase tumorigenicity in prostate cancer. Proc Natl Acad Sci U S A 102(3): 719-724.
- Pu X, Kamendulis LM, Klaunig JE (2006) Acrylonitrile-induced oxidative DNA damage in rat astrocytes. Environ Mol Mutagen 47(8): 631-638.
- Pu X, Kamendulis LM, Klaunig JE (2009) Acrylonitrile-induced oxidative stress and oxidative DNA damage in male Sprague-Dawley rats. Toxicol Sci 111(1): 64-71.
- Quast JF (2002) Two-year toxicity and oncogenicity study with acrylonitrile incorporated in the drinking water of rats. Toxicol Lett 132(3): 153-196.
- Suzuki H, Kumagai T, Goto A, Sugiura T (1998) Increase in intracellular hydrogen peroxide and upregulation of a nuclear respiratory gene evoked by impairment of mitochondrial electron transfer in human cells. Biochem Biophys Res Commun 249(2): 542-545.
- Wang D, Kreutzer DA, Essigmann JM (1998) Mutagenicity and repair of oxidative DNA damage: insights from studies using defined lesions. Mutat Res 400(1-2): 99-115.
- Yang Z, Schumaker LM, Egorin MJ, Zuhowski EG, Guo Z et al. (2006) Cisplatin preferentially binds mitochondrial DNA and voltage-dependent anion channel protein in the mitochondrial membrane of head and neck squamous cell carcinoma: possible role in apoptosis. Clin Cancer Res 12(19): 5817-5825.
- Zhou S, Kachhap S, Sun W, Wu G, Chuang A, et al. (2007) Frequency and phenotypic implications of mitochondrial DNA mutations in human squamous cell cancers of the head and neck. Proc Natl Acad Sci U S A 104(18):7540-7545.
- Zoratti M, Szabo I (1995) The mitochondrial permeability transition. Biochim Biophys Acta 1241(2): 139-176.





