Evaluation of Leukocyte DNA Damage and Antioxidant Defense in Graves’ Disease; Effect of Medical Treatment
Akcay T1*, Himmetoglu S1, Yasar O1, Erdem T2, Gundogdu S2, and Dincer Y1
1 Department of Biochemistry, Cerrahpasa Medical Faculty, Istanbul University, Istanbul, Turkey.
2 Department of Internal Medicine, Cerrahpasa Medical Faculty, Istanbul University, Istanbul, Turkey.
*Corresponding Author
Tulay Akcay,
Department of Biochemistry, Cerrahpasa Medical Faculty,
Istanbul University, Istanbul, Turkey.
Tel: +90 542 2161359
E-mail: tulakcay@yahoo.com
Received: May 28, 2017; Accepted: June 27, 2017; Published: June 30, 2017
Citation: Akcay T, Himmetoglu S, Yasar O, Erdem T, Gundogdu S, et al., (2017) Evaluation of Leukocyte DNA Damage and Antioxidant Defense in Graves’ Disease; Effect of Medical Treatment. Int J Clin Pharmacol Toxicol. 6(3), 280-283. doi: dx.doi.org/10.19070/2167-910X-1700046
Copyright: Akcay T© 2017. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
Abstract
Background: Oxidative stress has been implicated in many pathological conditions, including hyperthyroidism. In this study, our aim was to investigate the effect of medical treatment on oxidative/antioxidative status and subsequent DNA damage in Graves’ disease, in terms of reduced glutathione (GSH) levels, superoxide dismutase (SOD), glutathione peroxidase (GPx) activity and DNA strand breaks.
Methods: Fifty female patients suffering from Graves’ disease and thirty-seven healthy female controls were recruited in the study. Blood samples were taken from the patients before and after treatment. Free T3, free T4 and thyroid stimulating hormone (TSH) levels were determined using chemiluminescent particle assay, GSH, SOD and GPx activity using photometric techniques, and comet assay was utilized to analyze strand breaks with formamidopyrimidine DNA glycosylase (FPG) sensitive sites.
Results: Free T3, free T4, GPx activity, DNA strand breaks and FPG sensitive sites were significantly higher, and TSH and GSH levels were significantly lower in patients prior to treatment compared to healthy controls. Following treatment, the levels of free T3, free T4 and TSH were restored to those of healthy controls, but the rise in GSH levels and the drop in GPx activity, strand breaks and FPG sensitive sites were not as profound, though still significantly different than pre treatment levels.
Conclusions: The markers of oxidative stress and DNA damage are not restored as quickly as thyroid function tests in Graves’ disease following treatment, suggesting the oxidative damage caused by hyperthyroidism may have long term effects.
2.Introduction
3.Materials and Methods
4.Results
5.Discussion
6.Acknowledgments
7.References
Keywords
Hyperthyroidism; Graves’ Disease; Oxidative Stress; DNA Damage; Comet Assay.
Introduction
Graves’ disease is the most common cause of hyperthyroidism [1]. It is characterized by palpitations, fatigue, heat intolerance, weight loss, and sometimes dermopathy and ophthalmopathy manifests through the progression of the disease [2]. Thyroid hormones increase oxidative metabolism by affecting basal metabolic rate and several mitochondrial enzymes [3, 4], and hyperthyroidism causes changes in the antioxidant systems in various tissues by increasing the production of free radicals [5]. These radicals induce a variety of lesions in DNA, including DNA strand breaks, oxidized bases, and formation of cross-links between DNA and proteins [6]. DNA damage is constantly fixed by repair mechanisms in aerobic organisms; however, if cell division occurs before repairs are completed, the DNA damage becomes permanent [7]. A major site of radical attack is at the 8th position of guanine to yield 8-hydroxy-2-deoxyguanosine (8-OHdG). This modification might cause severe mutations [8], and sites within DNA which contain these alterations are more susceptible to strand breaks. These lesions can be detected by employing single cell gel electrophoresis, also known as comet assay. Furthermore, utilizing formamidopyrimidine DNA glycosylase (FPG), a damage specificrepair endonuclease, during this assay will cause additional strand breaks at sites with oxidized bases [9].
Several antioxidant defense mechanisms exist in order to counter the harmful effects of free radicals. One of these defense agents is glutathione [10]. Glutathione, in its reduced form (GSH), is a physiological component of the intracellular antioxidant mechanisms. It acts against the effects of free radicals through serving as a cofactor for the enzyme glutathione peroxidase (GPx) [11]. Beside GPx, superoxide dismutase (SOD) is another enzyme defending the organism against free radicals [11].
Overproduction of free radicals may exceed the capacity of antioxidant defense mechanisms, and this will give rise to the condition known as oxidative stress. This condition has been implicated in the pathogenesis of many diseases [12-16]. The current study evaluates the relationship between DNA damage, in terms of strand breaks, and antioxidant mechanisms, in terms of GSH, GPx and SOD, in patients with Graves’ disease.
Materials and Methods
Fifty untreated female patients suffering from Graves’ disease but not any other metabolic condition were enrolled in the study. The control group consisted of thirty-seven female volunteers. The participants of both the patient and control group did not smoke or receive any antioxidant drugs or vitamin supplements. The exclusion criteria also included hypertension, diabetes mellitus, hyperlipidemia, liver disease, renal disease, endocrine and immunological disorders for patients and controls. Included participants had no history of liver or renal disease, and all had normal liver and kidney function test results. The ethical committee at Cerrahpasa Medical Faculty approved this study and informed consent was obtained from each participant.
10 mL of venous blood was drawn into a heparinized tube from each patient and healthy volunteer. GSH levels were determined using the method by Beutler et al., as described before [17]. The activity of enzymes GPx and SOD were measured using spectrophotometric kits (Randox, UK). The strand breaks in DNA of leukocytes was assessed using Comet assay by Singh et al., as described before [18]. Heparinized blood was mixed 1.4% low melting point agarose in Dulbecco's buffer at 37°C and then this mixture was layered onto slides and covered with a coverslip at 4°C for at least 5 minutes. After removing the coverslips, the slides were submersed in lysing solution (2.5 M NaCl, 100 mM EDTA-Na2, 10mM Tris-HCl, 200mM NaOH, 1% triton X-100, and 10% DMSO) for 1 hour. Subsequently slides were immersed in alkaline electrophoresis buffer (pH ˃ 13) at 4°C for unwinding and electrophoresis (20V/400mA, 24 min). The loops of DNA extend towards the anode, giving an appearance of the tail of a comet when stained with ethidium bromide and viewed by fluorescence microscopy. In order to evaluate the degree of damage, comet images were scored visually. Each comet was classified to five categories (0 - 4) according to the extent of DNA migration. The comets with bright heads and no apparent tails were assigned to category 0. Images with very little heads and long diffused tails to category 4. Comets displaying features intermediate between category 0 and 4 were divided and assigned to easily distinguishable categories 1, 2 and 3. The number of comets in each category was counted and average DNA damage in case of strand breaks was expressed as arbitrary units (au) which is related to the percentage of DNA in the tail [19].
The oxidative DNA damage (8-oxo-Gua adducts) is measured as a strand break by using 8-oxo-Gua specific endonuclease. The assay was carried out both with and without using FPG, a bacterial protein which generates additional breaks at sites containing 8-oxo-Gua, for each sample. The net amount of base damage by FPG was obtained by subtracting the percent DNA in tail without FPG incubation from the percent DNA in tail with FPG incubation [20].
These laboratory procedures were performed at the Department of Biochemistry at Cerrahpasa Medical Faculty. For thyroid function tests, an additional 5 mL venous blood was drawn into a serum tube from every participant and the levels of free T3, free T4 and thyroid stimulating hormone (TSH) were determined using chemiluminescent particle assay at the Central Laboratory at Cerrahpasa Medical Faculty.
All patients only received methimazole (20 mg/day) and propylthiouracil (100 mg/day) as medical treatment. No alternative modality of treatment (surgery or radioactive iodine) were employed. Samples were taken a second time from those patients who visited for follow-up 6months after treatment. These blood samples were put through the same laboratory procedures in order to compare pre- and post treatment levels.
For statistical analysis, SPSS Statistics version 20 was used. Due to the distribution of data, groups were compared using Mann- Whitney’s U test. For correlations, Spearman’s rho was utilized. All data were evaluated within 95% confidence interval with a significance level at p<0.05.
Results
Among the controls, strand breaks appeared to increase moderately with age (rho: 0.659, p<0.01). However, when all participants (both controls and patients) were considered, this correlation was lost (rho: 0.176, p>0.05). Besides, there was no significant difference between the controls and patients in terms of age. Also within the controls, no parameter correlated with any other except GSH and GPx, though this correlation was weak (rho: 0.451, p<0.05).
The levels of each analyte measured in all groups are summarized in Table 1. Only 6 patients did not visit for follow-up. None of the groups differed in terms of age. Free T3, free T4, strand breaks, FPG sensitive sites and GPx were all significantly increased, while TSH and GSH were significantly decreased in patients prior to treatment compared to healthy controls (all p<0.001). Following treatment, all of these parameters approached the values of controls. Post treatment levels of free T3 and free T4 were significantly lower than pre treatment levels and there was no significant difference between post treatment levels and controls. In contrast, TSH levels were significantly increased following treatment, and no significant difference was observed between controls and post treatment levels. Strand breaks and GPx levels, on the other hand, were both significantly decreased following treatment, but were still significantly higher than controls. GSH levels were also significantly increased after treatment, but still significantly lower compared to controls nonetheless. FPG sites, however, did not decrease significantly following treatment. Finally, SOD did not differ between any groups.
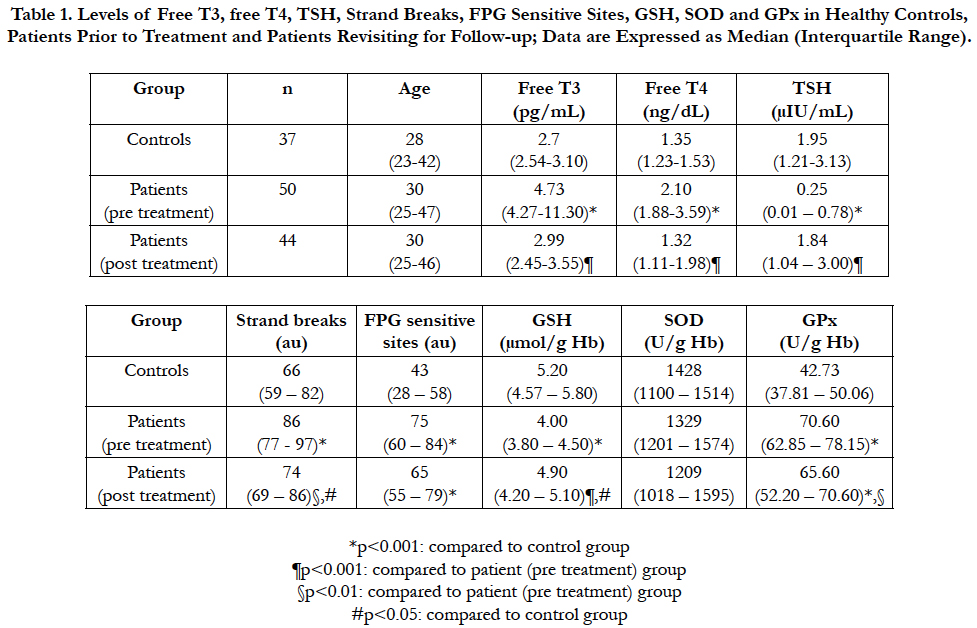

Table 1. Levels of Free T3, free T4, TSH, Strand Breaks, FPG Sensitive Sites, GSH, SOD and GPx in Healthy Controls, Patients Prior to Treatment and Patients Revisiting for Follow-up; Data are Expressed as Median (Interquartile Range).
Among the patients, free T3 and free T4 were moderately positively correlated with each other (rho: 0.693, p<0.01). In addition, TSH was moderately negatively correlated both with free T3 (rho: -0.636) and free T4 (rho: -0.531) (both p<0.01). Interestingly, TSH was also slightly correlated positively with GSH (rho: 0.357) and negatively with GPx (rho: -0.309) (both p<0.05). Moreover, GSH correlated negatively with free T4 (rho: -0.454, p<0.01). In contrast to the controls, the levels of GSH and GPx were negatively correlated with each other within patients (rho: -0.319, p<0.05).
When both controls and patients were considered, all correlations found within patients were still present, but also new correlations were observed. Within the whole study population, GPx were also positively correlated with free T3 (rho: 0.622), free T4 (rho: 0.522), strand breaks (rho: 0.353) and FPG sensitive sites (rho: 0.469). On the other hand, GSH was also negatively correlated with free T3 (rho: -0.522) and FPG sensitive sites (rho: -0.357). Finally, FPG sensitive sites displayed a slight negative correlation with TSH (rho: -0.443) (all p<0.01).
Discussion
Oxidative stress is thought to play a significant role in the pathogenesis of many diseases, including hyperthyroidism [21]. In Graves’ disease, elevated concentrations of thyroid hormones increase the basal metabolic rate, which increases oxygen consumption. This, in turn, accelerates the production of reactive oxygen species and free radicals [22]. To counter this, the human body possesses several antioxidant mechanisms.
One of the main endogenous antioxidants is glutathione. Its protective effects against free radicals have been widely studied [23]. The levels of its reduced form have also been investigated in patients with hyperthyroidism, though there are conflicting results. While some demonstrate its levels are decreased in hyperthyroidism and elevated after medical treatment [24], some report the opposite [25]. Our results support the former, as we can speculate that due to oxidative demand, GSH is depleted at a rapid rate in order to neutralize the harmful effects of free radicals generated as a result of excessive thyroid function.
GPx is an enzyme which uses GSH as its cofactor. In response to oxidative stress, its activity increases, and following treatment, the overproduction of free radicals by thyroid hormones is reduced, which in turn lowers its activity, as our results indicate. Some studies support our results [26, 27] while others indicate decreased levels before medical treatment [28, 29]. This decrease, however, has been attributed to nutritional state of the patients [22, 29].
As another antioxidant enzyme, SOD has also been investigated in Graves’ disease. While there have been some which report decreased activity [29], most studies demonstrate increased levels [26, 30, 31]. Our results support neither view, as we could not find any significant change in plasma SOD levels in patients compared to controls.
There have been a limited number of studies assessing DNA damage by comet assay in hyperthyroidism. In a recent animal study, it has been demonstrated that supraphysiological doses of thyorid hormones cause overproduction of reactive oxygen species resulting in increased DNA damage [32]. These findings support our results, as strand breaks and FPG sensitive sites were more abundant in our patient group compared to our controls. Another study involving humans investigated lymphocyte DNA damage in patients with Graves’ disease in vitro. In this study by Tang et al., [33], it was demonstrated that DNA damage was found to be increased in the lymphocytes of patients with Graves’ disease, and with antioxidant treatment of these lymphocytes in vitro, this damage was reduced to a varying degree.
According to our search of the medical literature, this is the first study evaluating oxidative stress in terms of GSH, SOD and GPx together with assessing leukocyte DNA damage by comet assay in Graves’ disease before and after treatment with antithyroid drugs. As our results indicate, GSH is reduced in Graves’ disease by increased activity of GPx. This leaves DNA to be more susceptible to oxidative damage, and thus to mutations. Antithyroid drugs lessen this effect to some degree, by antagonizing the effects of thyroid hormones. Following treatment, the levels of oxidative stress and DNA damage parameters approached those of healthy controls, but were still significantly higher. This is in contrast to studies by Adali et al., [24] and Komosinska-Vassev et al., [26]. There was no significant difference between post treatment and control levels in terms of GSH in the study by Adali et al, nor in terms of GPx in the study by Komosinska-Vassev et al., Nevertheless in our study, thyroid function test results did not significantly differ between controls and patients following treatment. This leads us to suggest that despite the return of thyroid function to normal levels, the oxidative damage, especially to the DNA, is not resolved as quickly.
The major limitations of our study are the relatively small sample size of groups, discontinued follow-up of patients, and the short span of the study being insufficient to observe the late complications of the disease. In future studies with larger sample sizes, the follow-up of patients can be extended in order to further investigate the relationship between oxidative DNA damage and the complications of Graves’ disease.
Acknowledgments
This work was supported by Scientific Research Projects Coordination Unit of Istanbul University.
References
- Burch HB, Cooper DS (2015) Management of Graves Disease: A Review. JAMA 314(23): 2544-2554.
- Smith TJ, Hegedüs L (2016) Graves' Disease. N Engl J Med. 375(16): 1552- 1565.
- Pereira B, Rosa LF, Safi DA, Bechara EJ, Curi R (1994) Control of superoxide dismutase, catalase and glutathione peroxidase activities in rat lymphoid organs by thyroid hormones. J Endocrinol. 140(1): 73-77.
- Videla LA, Sir T, Wolff C (1988) Increased lipid peroxidation in hyperthyroid patients: suppression by propylthiouracil treatment. Free Radic Res Commun. 5(1): 1-10.
- Asayama K, Kato K (1990) Oxidative muscular injury and its relevance to hyperthyroidism. Free Radic Biol Med. 8(3): 293-303.
- Shigenaga MK, Ames BN (1991) Assays for 8-hydroxy-2'-deoxyguanosine: a biomarker of in vivo oxidative DNA damage. Free Radic Biol Med. 10(3- 4): 211-216.
- Nelson DL, Cox MM (2013) Lehninger Principles of Biochemistry. (6th Edn), W. H. Freeman, New York.
- Cheng KC, Cahill DS, Kasai H, Nishimura S, Loeb LA (1992) 8-Hydroxyguanine, an abundant form of oxidative DNA damage, causes G----T and A----C substitutions. J Biol Chem. 267(1): 166-172.
- Kassie F, Parzefall W, Knasmüller S (2000) Single cell gel electrophoresis assay: a new technique for human biomonitoring studies. Mutat Res. 463(1): 13-31.
- Pompella A, Visvikis A, Paolicchi A, De Tata V, Casini AF (2003) The changing faces of glutathione, a cellular protagonist. Biochem Pharmacol. 66(8): 1499-1503.
- Muller FL, Lustgarten MS, Jang Y, Richardson A, Van Remmen H (2007) Trends in oxidative aging theories. Free Radic Biol Med. 43(4): 477-503.
- Aly DG, Shahin RS (2010) Oxidative stress in lichen planus. Acta Dermatovenerol Alp Pannonica Adriat. 19(1): 3-11.
- Halliwell B (2012) Free radicals and antioxidants: updating a personal view. Nutr Rev. 70(5): 257-265.
- Hwang O (2013) Role of oxidative stress in Parkinson's disease. Exp Neurobiol. 22(1): 11-17.
- Pohanka M (2013) Role of oxidative stress in infectious diseases. A review. Folia Microbiol (Praha). 58(6): 503-513.
- Santilli F, D'Ardes D, Davì G (2015) Oxidative stress in chronic vascular disease: From prediction to prevention. Vascul Pharmacol. 74: 23-37.
- Hasbal C, Aksu BY, Himmetoglu S, Dincer Y, Koc EE, et al., (2010) DNA damage and glutathione level in children with asthma bronchiale: effect of antiasthmatic therapy. Pediatr Allergy Immunol. 21(4): e674-678.
- Aksu BY, Hasbal C, Himmetoglu S, Dincer Y, Koc EE, et al. (2010) Leukocyte DNA damage in children with iron deficiency anemia: effect of iron supplementation. Eur J Pediatr. 169(8): 951-956.
- Collins AR, Ma AG, Duthie SJ (1995) The kinetics of repair of oxidative DNA damage (strand breaks and oxidised pyrimidines) in human cells. Mutat Res. 336(1): 69-77.
- Collins AR, Raslová K, Somorovská M, Petrovská H, Ondrusová A, et al., (1998) DNA damage in diabetes: correlation with a clinical marker. Free Radic Biol Med. 25(3): 373-377.
- Owczarek T, Kowalczyk E, Poliwczak AR, Bała A, Broncel M (2012) The evaluation of selected oxidative stress parameters in patients after radioiodine treatment of hyperthyroidism. Pol Merkur Lekarski. 32(192): 382-387.
- Vrca VB, Skreb F, Cepelak I, Romic Z, Mayer L (2004) Supplementation with antioxidants in the treatment of Graves' disease; the effect on glutathione peroxidase activity and concentration of selenium. Clin Chim Acta. 341(1-2): 55-63.
- Gebicki JM (2016) Oxidative stress, free radicals and protein peroxides. Arch Biochem Biophys. 595: 33-39.
- Adali M, Inal-Erden M, Akalin A, Efe B (1999) Effects of propylthiouracil, propranolol, and vitamin E on lipid peroxidation and antioxidant status in hyperthyroid patients. Clin Biochem. 32(5): 363-367.
- Seven A, Tasan E, Hatemi H, Burcak G (1999) The impact of propylthiouracil therapy on lipid peroxidation and antioxidant status parameters in hyperthyroid patients. Acta Med Okayama. 53(1): 27-30.
- Komosinska-Vassev K, Olczyk K, Kucharz EJ, Marcisz C, Winsz-Szczotka K, et al., (2000) Free radical activity and antioxidant defense mechanisms in patients with hyperthyroidism due to Graves' disease during therapy. Clin Chim Acta. 300(1-2): 107-117.
- Rybus-Kalinowska B, Zwirska-Korczala K, Kalinowski M, Kukla M, Birkner E, et al., (2008) Activity of antioxidative enzymes and concentration of malondialdehyde as oxidative status markers in women with newly diagnosed Graves-Basedow disease and after thiamazole therapy leading to euthyroidism. Pol Arch Med Wewn. 118(7-8): 420-425.
- Bednarek J, Wysocki H, Sowiński J (2004) Peripheral parameters of oxidative stress in patients with infiltrative Graves' ophthalmopathy treated with corticosteroids. Immunol Lett. 93(2-3): 227-232.
- Kaur A, Pandey S, Kumar S, Mehdi AA, Mishra A (2010) Oxidative stress profile in graves' ophthalmopathy in Indian patients. Orbit. 29(2): 97-101.
- Bednarek J, Wysocki H, Sowinski J (2004) Oxidation products and antioxidant markers in plasma of patients with Graves' disease and toxic multinodular goiter: effect of methimazole treatment. Free Radic Res. 38(6): 659-664.
- Lassoued S, Mseddi M, Mnif F, Abid M, Guermazi F, et al., (2010) A comparative study of the oxidative profile in Graves' disease, Hashimoto's thyroiditis, and papillary thyroid cancer. Biol Trace Elem Res. 138(1-3): 107-115.
- De Sibio MT, Luvizotto RA, Olimpio RM, Corrêa CR, Marino J, et al., (2013) A comparative genotoxicity study of a supraphysiological dose of triiodothyronine (T₃) in obese rats subjected to either calorie-restricted diet or hyperthyroidism. PLoS One. 8(2): e56913.
- Tang XL, Liu XJ, Sun WM, Zhao J, Zheng RL (2005) Oxidative stress in Graves' disease patients and antioxidant protection against lymphocytes DNA damage in vitro. Pharmazie 60(9): 696-700.





