Drug Design And Analysis InSilico of Sapelenin G, an Acyclic Triterpenoid as Potential Anti-Inflammatory
Ngabireng Marie. Claude1*, Menye Cyrille2, Kouam F.Simeon3, Ntede N .Hyppolite4, Tagoudjeu Jacques1, Awono Onana1
1 National Advanced School of Engineering/Department of Mathematics and Physical Science, University of Yaounde I, Yaounde, Cameroon
2 Faculty of sciences/Department of Physics, University of Yaounde I, Yaounde, Cameroon
3 Higher Teachers’ Training College/Department of Chemistry, University of Yaounde I, Yaounde, Cameroon
4 National Advanced School of Engineering/Department of Mechanics and materials, University of Yaounde I,Yaounde, Cameroon
*Corresponding Author
Ngabireng Marie. Claude
National Advanced School of Engineering/Department of Mathematics and Physical Science
University of Yaounde I, Cameroon.
E-mail: cngabire@yahoo.fr
Article Type: Research Article
Received: June 06, 2013; Accepted: June 24, 2013; Published: July 27, 2013
Citation: Ngabireng Marie. Claude, et al., (2013) Drug Design and Analysis In Silico Of Sapelenin G, An Acyclic Triterpenoid as Potential Anti-Inflammatory. Int J Clin Pharmacol Toxicol. 2(4), 66-72. doi: dx.doi.org/10.19070/2167-910X-1300014
Copyright: Ngabireng Marie. Claude© 2013. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
Abstract
Diverse non-steriodal anti-inflammatory drugs and COX-2 inhibitors are a class of drugs which selectively inhibit COX-2, provide relief from pain and inflammation. However, they lack anti-thrombotic activity and hence lead to cardiovascular and renal liabilities apart from gastrointestinal irritation. To ameliorate this situation, research can be foccuss on the products originating from natural products that could offer better relief from inflammation than the currently used commercial drugs. Aspirin blocks the cyclooxygenase enzyme (COX 1, 2) which is involved in the ring closure and the addition of O2 to arachidonic acid, converting it to prostaglandins (which induce inflammation, pain and fever). The present study is undertaken to analyse the docking efficacy of aspirin with the target molecule (2AW1), to assess the best ligand for inhibiting COX and to analyze the docking program by Arguslab. Substituting aspirin ligand by sapelenin G, the finding suggests that sapelenin G is a better ligand than aspirin. It satisfies Lipinski Rule of 5. The PASS (Prediction of Activity Spectra for Substances) prediction results show that Sapelenin G has an anti-inflammatory activity. Toxicity estimations of Sapelenin G using Toxtree on humans and based on the Cramer rules, Verhaar, Structural Alerts for Reactivity in Toxtree (START) biodegradability, eye irritation/corrosion and skin irritation/corrosion fell into class 3, 5, 1, 2 and 1, respectively. Application of the Benigni-Bossa method showed that this compound is negative for genotoxic carcinogenicity and negative for nongenotoxic carcinogenicity. The cytochrome P-450 mediated drug metabolism is negative for Sapelenin G only in SMARTCyp.Rank2.sites of metabolism, and it fell into unreactive group of compounds by Michael addition. A skin sensitization evaluation reveals that the compound has no skin sensitization alert identified, moreover, Kroes Threshold of Toxicological Concern (TTC) decision tree reveals that the Substance would not be expected to be a safety concern.
2.Introduction
3.Materials and Methods
3.1.Molecular docking
3.2.ADME/toxicity testing
3.3.Prediction Activity Spectra for Substances (PASS)
3.4.Toxtree
4.Results and Discussion
5.Acknowledgment
6.References
Keywords
Sapelenin G, Cyclooxygenase, Docking, Anti-inflammatory activity.
Introduction
Sapelenin G, an acyclic triterpenoid, known under its UIPAC name as (10E, 14E, 18E)-2 ,6,10,15,19,23-hexamethyltetracosa-10,14,18-triene-2,3,6,7,22,23-hexol (fig.1b) is a molecule extracted from a sapele tree scientifically known as Entadrophragma cylindricum [1]. It has been reported that triterpenes possess many useful properties among which anti-inflammatory [1, 2]. This activity is manifested by their interaction with a number of targets [3]; including nitric oxide synthase (iNOS), nuclear factor-kB (NF-kB), heme oxygenase (HO) and cyclooxygenase (COX).
The latest, also known as prostaglandin H Synthase (PGH synthase /PGHS/PHS) is a prominent and well-studied protein which catalyzes the conversion of arachidonic acid to prostaglandin H2 (PGH2), the committed step in prostaglandin (PG) biosynthesis. There are two isoforms of this enzyme: cyclooxygenase 1 (COX1) and cyclooxygenase 2 (COX2). COX1 are responsible for the maintenance and the protection of the gastrointestinal tract, COX2 is responsible for inflammation and pain [4].
The existing non-steroidal and anti-inflammatory drugs (NSAIDs) differ in their relative specificities for COX-1 and COX-2; while aspirin is equipotent at inhibiting COX-2 and COX-1 enzymes in vitro [5].
The first crystal structure of human cyclooxygenase-2, in the presence of selective inhibitor, is similar to that of cyclooxygenase-1; COX-1 and COX-2 are homodimers. The structure of NSAID binding site is also well conserved, although there are differences in its overall size. COX-1 and COX-2 are both targets of nonselective nonsteroidal anti-inflammatory drugs including aspirin and ibuprofen, while COX-2 activity is selectively blocked by COX-2 inhibitors called coxibs (e.g.,celecocib). Aspirin is a unique inhibitor that causes a time-dependent, covalent inhibition.
In this study, we are reporting probable binding mechanism of sapelenin G with COX by molecular docking. This molecule shows a good binding to COX than aspirin and has drug-like properties.
Materials and Methods
In the field of molecular modeling, docking is a method which predicts the preferred orientation of one molecule to another when bound to each other form a stable complex. Docking is frequently used to predict the binding orientation of small molecule drug candidates to their protein targets in order to predict the affinity and activity of the small molecule. Docking plays an important role in the rational design of drug.
In this study, the ligand aspirin will be docked with a receptor molecule using softwares and bioinformatic tools. The nonselective COX inhibitor aspirin, a clinically used anti-inflammatory agent, was used as the reference standard in this study and the general procedure for in silico analysis is detailed as follow: The chemical structures for ligands were drawn using Chemaxon/ Marvin Sketch, the generated structures were converted to 3D and minimized, the target COX protein (PDB ID: 2AW1) was retrieved from the RCSB Protein Data Bank as a PDB file and normalized, the ligands were docked onto entry 2AW1 of Brookhaven Protein Data Bank (PDB), the docking was performed using the robust Arguslab Molecular Modeling Program.
ADME (absorption, distribution, metabolism, and excretion) determines drug like activity of the ligand molecules based on Lipinski Rule of 5[6]. Increasing clinical failures of new drugs call for a more effective use of ADME/TOX technologies, becoming more advanced and reliable in terms of accuracy and predictiveness, an increase in their usage is expected during the initial development and screening phase of innovative drugs. [7].
This computer system can predict biological activity based on structural formula of a chemical compound. The PASS approach is based on the suggestion, Activity=Function (Structure).Thus, “comparing” structure of a new substance with that of the standard biologically active substances, it is possible to find out whether a new substance has a particular effect or not. PASS estimates the probabilities of a particular substances belonging to the active and inactive sub-sets from the SAR Base (Structure-Activity Relationships Base)[8].
PASS uses Sdfile (.sdf) or MOLfile (.mol) formats as an external source of structure and activity data to prepare both SAR Base and the set of substances to be predicted[9]. SD files can be exported either from ISIS/Base 2.0+ (MDL Information Systems, Inc.) or from another molecular editor which has the option of SD file’s export. MOLfiles can be prepared by ISIS/ Draw or by Marvin Sketch.
The result of prediction is returned in the form of a table containing the list of biological activity with the appropriate probability values (i.e) the values defining the likelihood for a given activity type to be either revealed PASS Activity(Pa) or not revealed PASS Inactivity (Pi) for each activity type from the predicted biological activity spectrum. Their values vary from 0.000 to 1.000. Only those activity types for which Pa > Pi are considered possible.
Toxtree was developed by IDEA consult. Ltd. (Sofia, Bulgaria). It has been made available as a free download [10]. The current version (v.2.2.0 October, 2010) includes decision trees for predicting Cramer rules, Cramer rules with extensions, Verhaar scheme, START biodegradability, eye irritation and corrosion, structure alerts for the in vivo micronucleus assay in rodents, Michael acceptors, Benigni/Bossa rulebase (for mutagenicity and carcinogenicity), skin irritation/skin corrosion, cytochrome P450-mediated drug metabolism, skin sensitisation alerts and Kroes TTC decision tree. The main window of Toxtree is classified into three different areas: the compound properties area, used to resume the available information on the current compound; the compound structure diagram area which shows a picture of the current compound and provides an easy way to navigate through the list of compounds in the currently opened file and the classification area which provides access to the classification output for the current compound.
To generate predictions with Toxtree, user-defined molecular structures can be input as SMILES codes or using the built-in 2D structure diagram editor. The software can also be used to perform batch processing of large numbers of compounds by importing datasets of various file types.
Results and Discussion
Molecular docking of ligands with the target proteins are routinely and extensively used to reduced cost and time of drug discovery. The target 2AW1 is docked with the geometrically optimized ligands: Aspirin and Sapelenin G. The two compounds showed very good interaction energies and the best ligands pose were tabulated (fig 2,3 table 1). Among the two compounds, Sapelenin G has a highly negative energy, with an interaction energy (-10.77 Kcal/mol), it has a good binding interaction than aspirin (with an interaction energy -9.24 Kcal/mol). The acyclic triterpenoid interact with Leu-141 and the OH group, this is responsible for tight binding with COX. The interatomic distance of hydrogen bonding between OH and Leu-141 is 2.566 Å.
The drug like activity of the ligand molecules are characterized using ADME properties. sapelenin G satisfy Lipinski rule of 5 and ADME properties results are shown in Table 2.
The prediction of activity spectra for substances (PASS) predicts the biological activity spectrum (BAS) [11] of a compound represents the complex of pharmacological and physiological effects and biochemical mechanisms of actions, specific toxicity for a compound on the basis of its structural formula [12].
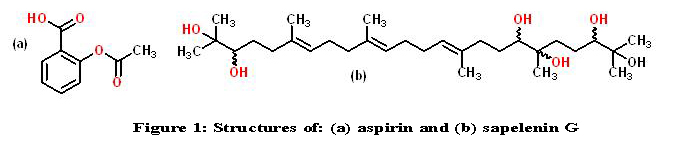
Figure 1: Structures of: (a) aspirin and (b) sapelenin G.
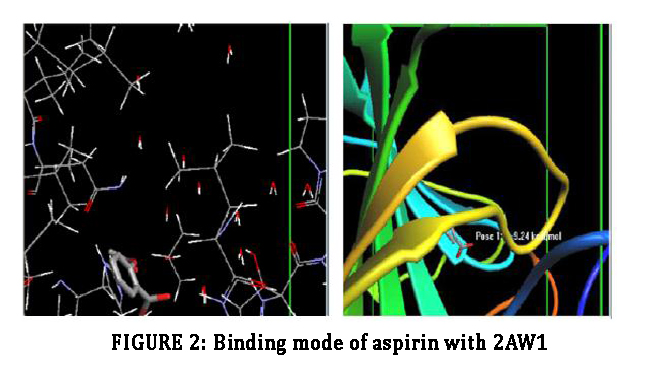
Figure 2: Binding mode of aspirin with 2AW1.
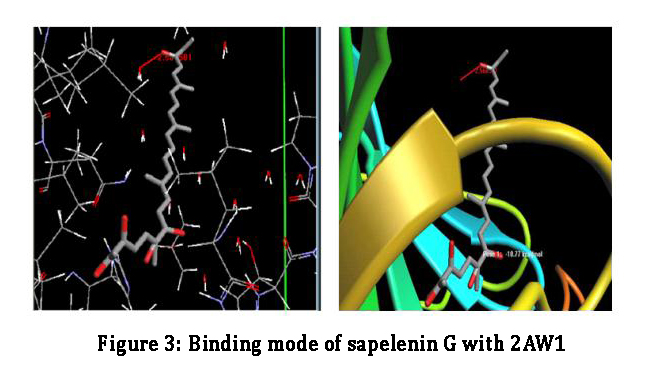
Figure 3: Binding mode of sapelenin G with 2AW1.
In our study, for Pa>Pi, PASS predicts 590 and 322 biological activity for aspirin and sapelenin G respectively. If we increase the threshold value up to Pa > 30; 50 and 70%, the number of activities is: 235 and 108, 106 and 50, 32 and 11, respectively for aspirin and sapelenin G. (table 3). Among the predicted biological activities when Pa>70%, we can note that our two molecules possess all one anti-inflammatory activity, the aspirin possesses a probability of presence of the anti-inflammatory activity valued to76.50 and the sapelenin G possesses a probability of 73.20. In the table 5, we presented 22 activities of the aspirin molecule out of the 33 well stocked by the PASS for Pa>70%, for reasons of spaces and without forgetting however that the goal here is to show that our two molecules possess the anti-inflammatory activities. We can read it in line 9 in the table 4 and in line 20 in the table 5. (Table 4, Table 5).


Table 1: Arguslab docking scores for aspirin and sapelenin G.
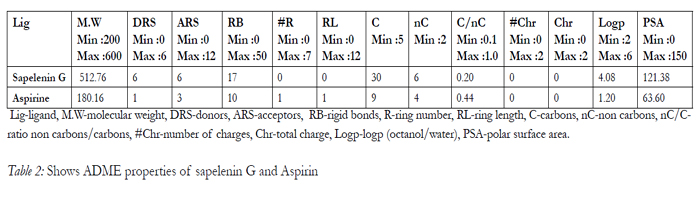
Table 2: Shows ADME properties of sapelenin G and Aspirin.
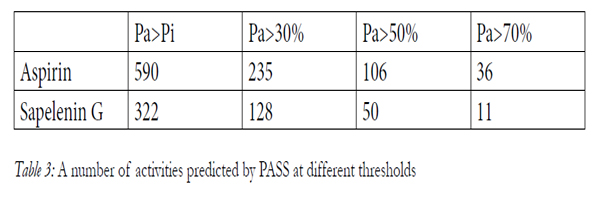
Table 3: A number of activities predicted by PASS at different thresholds.
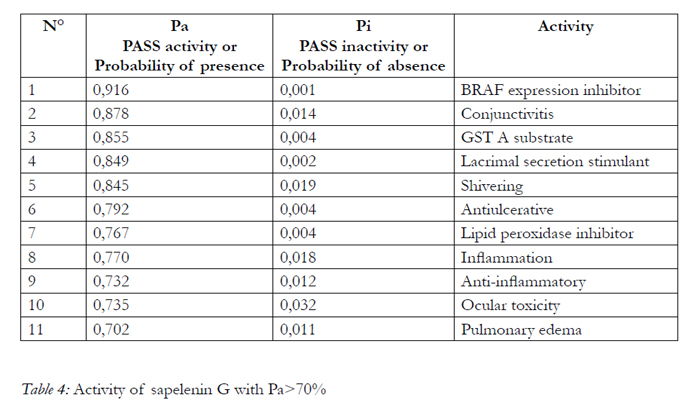
Table 4: Activity of sapelenin G with Pa>70%.
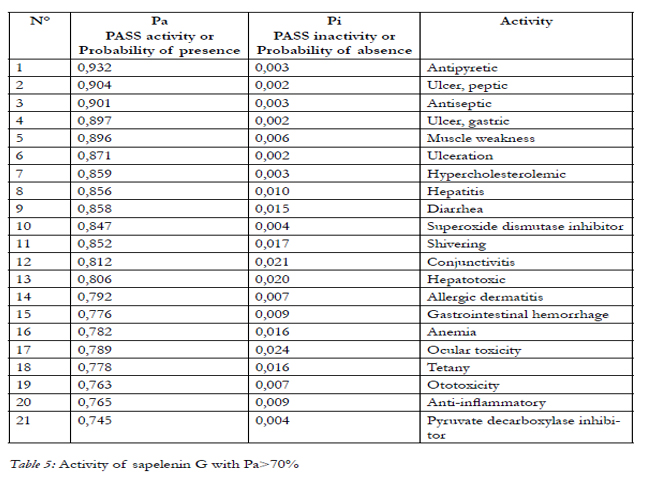
Table 5: Activity of sapelenin G with Pa>70%.
Toxtree is able to estimate toxic hazard by applying a decision tree approach. The decision tree approaches and classes were then used to predict the toxicities (Table 6, 7).
Cramer rules classify chemicals into three structural classes based on a decision tree. Questions asses different features include structural features (functional groups, ring substituent, etc.), propensity of reaction, natural occurrence in body and in traditional foods and the logic of tree relies primarily on knowledge of common metabolic pathways [13, 14].
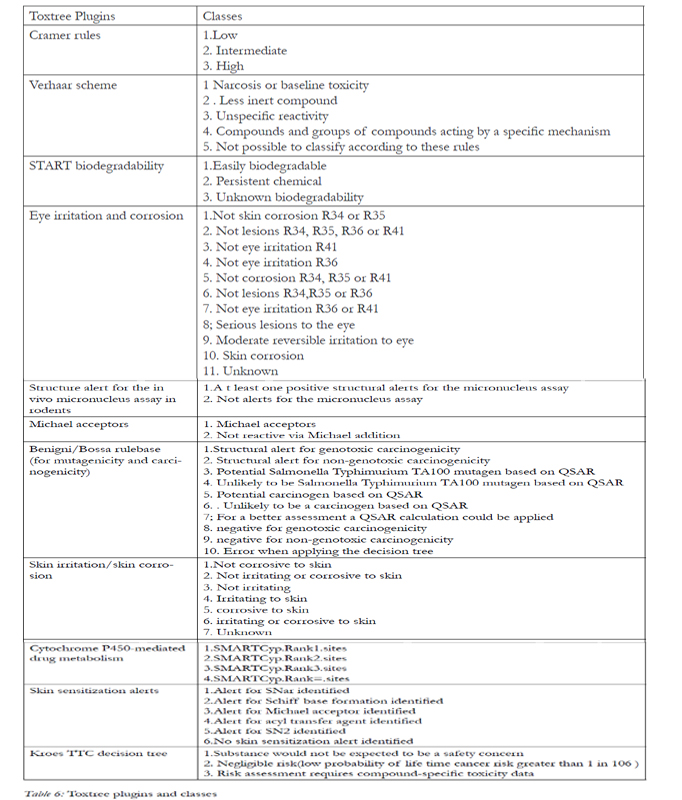
Table 6: Toxtree plugins and classes

Table 7: Toxic hazard classification as predicted by Toxtree
As shown in Table 7, Aspirin fell into class 1 and sapelenin G fell into class 3, indicating that the compound is a substance that permit no strong initial impression of safety and may even suggest a significant toxicity. The sapelenin G compound fell into class 3 by Cramer rules because a negative response to question 1 “Normal constituent of the body” due to Open chain in the compounds.
Potential mechanisms of toxic action were identified for these compounds through application of the Verhaar scheme. A number of mechanisms have been identified that can lead to aquatic toxicity, with the majority of industrial chemicals exerting their toxic influence via two non-covalent mechanisms; polar narcosis and non-polar narcosis. According to the Verhaar scheme, sapelenin G fell into class 5 and aspirin fell into class 3 (Table 7). Compounds which cannot be classified as belonging to classes 1, 2 or 3 and that are not known to be compounds acting by a specific mechanism can only be classified as ‘not possible to be classified according to these rules’.
START biodegradation and persistence is a compilation of structural alerts for environmental persistence and biodegradability. These structural alerts are molecular functional groups or substructures that are known to be linked to the environmental persistence or biodegradability of chemicals. According to START biodegradability, all of the compounds fell into class 1 because they have alcohols function (Table 7).
Skin irritation and skin corrosion refer to localized toxic effects resulting from a topical exposure of the skin to a substance. There is strong evidence that chemicals which are corrosive to the skin should also be classified as being corrosive to the eye. Toxtree implements the BfR rules (The German Federal Institute for Risk Assessment) for predicting skin/eye irritation and corrosion The system is based on the combined use of two predictive approaches: exclusion rules based on physicochemical cut-off values to identify chemicals that do not exhibit a certain hazard (e.g., skin irritation/ corrosion) and inclusion rules based on structural alerts to identify chemicals that do show a particular toxic potential. According to eye irritation/corrosion, aspirin fell into class 11 “unknown” , sapelenin G fell into class 2 “Not lesions R34, R35, R36 or R41” and for skin irritation/corrosion, aspirin is classified as irritating to skin, sapelenin G is classified as not corrosive to skin (table 7 ).
Structure alerts for the micronucleus assay in rodent resulted in a classification of class 1 for all compounds (Table 7). It means that all of compounds have at least one positive alert for the micronucleus assay. This plugin provides a list of structural alerts for a preliminary screening of potential in vivo mutagens. These structural alerts are molecular functional groups or substructures that are known to be linked to a positive in vivo micronucleus assay.Benigni-Bossa method showed that the two compounds (table7) are negative for genotoxic carcinogenicity and negative for nongenotoxic carcinogenicity.
Michael acceptors resulted in a classification of class 2 for all compounds, because they don’t have any structure alert for Michael acceptor.( table7).
Toxtree can identify potential mechanism of toxic action for skin sensitization. The sapelenin G was identified as No skin sensitization alert identified and aspirin is classified as Alert for acyl transfer agent identified. (Table 7)
Cytochrome P450-mediated drug metabolism resulted in positive for all site of metabolism except for sapelenin G which is negative in SMARTCyp.Rank2. sites. SMARTCyp is an in silico method that predicts the sites of metabolism for drug metabolites mediated by cytochrome P450 3A4 isoform. The idea behind SMARTCyp is that activation energies of CYPs reacting with ligand fragments computed by quantum chemical methods are the best possible reference for the reactivity of a fragment. Those results indicated that the compounds were likely easily metabolized. TTC is a pragmatic risk assessment tool which is based on the principle of establishing a human exposure threshold value for all chemicals, below which there is a very low probability of an appreciable risk to human health. Kroes TTC decision tree showed that the Substance would not be expected to be a safety concern for the two compounds (Table 7).
The classification numbers refer to Table 6.*Structure alerts for reactivity in Toxtree (START) biodegradability, #Structure alerts for the in vivo micronucleus assay in rodents, +Benigni/Bossa rule base for mutagenicity and carcinogenicity, ×Cytochrome P450-Mediated drug Metabolism, ∞Kroes threshold of toxicological concern (TTC).
The results of the present study have clearly demonstrated that sapelenin G possess anti-inflammatory activity, identified with the help of molecular docking and prediction of activity spectra for substance (PASS), the PASS reveals that sapelenin G possess eleven activities, with Pa>70% (table 2), among them, anti-inflammatory activity, with Pa = 73.20% (table 4). The drug like activity of sapelenin G is characterized using ADME properties, sapelenin G satisfy Lipinski rule of 5. Different type of toxicological hazard predicted by Toxtree reveals that sapelenin G could be categorized as the substance could not be expected to be safety concern (by TTC) and not corrosive to skin. The compound is likely easily metabolized, except in SMARTCyp.Kank2.sites. It showed at least, one positive alert for micronucleus assay. The sapelenin G may be considered as novel inhibitor of COX and as promising lead-compound for developing new anti-inflammatory drug.
Acknowledgment
Part of this work was supported by the International Foundation of Science (No. F/4893-1) and the Third World Academy Science (No 10-004 RG/CHE/AF/ AC-I). SKF also thank the Humboldt foundation for equipment donation.
References
- Kouam SF, Kusari S, Lamshöft M, Tatuedom OK, Spiteller M (2012) Sapelenins G-J, acyclic triterpenoids with strong anti-inflammatory activities from the bark of the Cameroonian medicinal plant Entandrophragma cylindricum. Phytochemistry 83: 79-86
- Manikrao AM, Khatale PN, Jawarkar RD, Vyas JV, Mahajan DT et al. (2011) Presuming the probable Anti-inflammatory Mechanism of ursolic acid: a plant derived pentacyclic triterpenoid, using molecular docking. J.Comput. Mol.Design 1(2): 9-13
- Kidd BL, Urban LA (2001) Mechanism of anti-inflammatory pain. Br.J. Anaesth. 87(1):3-11
- Goodsell DS (2000) The Molecular Perspective: Cyclooxygenase-2. Oncologist 5: 169-171.
- Debyani S, Sahu RK (2010) Drug designing and docking efficacy assessment of halogen substituted aspirin. Researcher 2(10):17-23
- Konstantin VB, Yan AI, Nikolay PS, Andrey AI, Sean E (2005) Comprehensive computational assessment of ADME properties using mapping techniques. Curr. Drug Discov. Technol 2: 99-113.
- Rani G J, Vinoth M, Anitha P (2011) Molecular docking studies on oxidosqualene cyclase with 4-piperidinopyridine and 4-piperidinopyrimidine as its inhibitors. J. Bioinform. Seq. Anal. 3(3): 31–36
- Poroikov VV, Filimonov D (2000) Medicinal Chemistry, Bologna (Italy). Abstr. XVIth Intern. Symp.
- http://www.mdli.com
- http://toxtree.sourceforge.net/download.html#Toxtree_2.2.0
- Soheila A, Gerhard B, Bertram C, Michael K, Dmitri F, Vladimir P (2001) Discriminating between drugs and non drugs by prediction of activity spectra for substances (PASS). J. Med. Chem 44: 2432- 2437
- Poroikov VV, Filimonov DA, Yu VB, Lagunin AA, Kos A (2000) Robustness of biological activity spectra predicting by computer program PASS for noncongeneric sets of chemical compounds. J. Chem. Inf. Comput. Sci. 40(6): 1349-1355
- Patlewicz, G, Jeliazkova N, Safford R.J., Worth AP, Aleksiev B (2008) An evaluation of the implementation of the cramer classification scheme in the toxtree software. SAR and QSAR Environ. Res. 19: 495-524.
- Asmiyenti DD, Kartasasmita RE, Slamet I, Daryono HT (2012) Toxicity Prediction of Photosensitizers Bearing Carboxylic Acid Groups by ECOSAR and Toxtree. J Pharmacol Toxicol 7: 219-230.





