Rare Presentation of a Rare Orthopedic Pathology: Erdheim Chester Disease
Samona J*, Owen J, Martin S
McLaren Flint Regional Medical Center, Orthopedic Surgery Division, Flint, Michigan, S. Ballenger Highway, Flint, MI, USA.
*Corresponding Author
Jason Samona DO,
Orthopedic Surgery, McLaren Regional Medical Center,
Orthopaedic Surgery Department, 401 S. Ballenger Highway, Flint, MI 48532, USA.
Tel: 248.939.6263
Fax: 810.342.2150
E-mail: Jasonsamona@yahoo.com
Received: October 04, 2016; Accepted: March 22, 2017; Published: March 24, 2017
Citation: Samona J, Owen J, Martin S (2017) Rare Presentation of a Rare Orthopedic Pathology: Erdheim Chester Disease. Int J Chronic Dis Ther. 3(1), 60-62. doi: dx.doi.org/10.19070/2572-7613-1700011
Copyright: Samona J© 2017. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
Abstract
In 1930, William Chester and pathologist Jakob Erdheim discovered the pathology currently known as Erdheim Chester disease. There are only approximately 249 histologically confirmed patients to have been diagnosed with this disease as noted in the medical literature. We describe a case report of an indolent form of Erdheim Chester Disease (ECD), in a patient in whom the disease has been present for approximately 14 years. Despite widespread sclerosis of long bones in the extremities, the patient has no complaints of peripheral bone pain. Although, the patient does have involvement of the jaw with subsequent loss of multiple teeth secondary to the facial manifestations of the disease. This is also the first patient have the disease incidentally discovered after an ACL injury, and the first ECD patient to receive an ACL reconstruction.
2.Case Report
3.Discussion
4.References
Introduction
In 1930, William Chester and pathologist Jakob Erdheim discovered the pathology currently known as Erdheim Chester disease. There are only approximately 249 histologically confirmed patients to have been diagnosed with this disease as noted in the medical literature [1]. This rare systemic inflammatory disease confuses pathologists, in that there are characteristics which are both neoplastic and non-neoplastic in nature. ECD is not classified as a cancer, autoimmune disorder, storage disorder, infectious disease, or any other clear pathological entity. It is characterized as both a Non-Langerhans cell histiocytosis; however, it may coexist with Langerhans histiocytosis. Inflammatory infiltrates are noted on pathological specimens, radiologically sclerosis of long bones may be noted, as well as involvement of the jaw in rare cases. Radio-nucleotide scans show high metabolic activity in these areas of sclerosis, most notably in the bilateral femurs in most patients.
The origins of this disease are unclear, although recently there has been genetic analysis which reveals association with chromosomal translocation t(12;15;20) (q11;q24;p13.3), and somatic BRAF gene mutations. These findings are preliminary in nature, and further genetic analysis is needed to better characterize the disease. ECD mostly affects middle-aged men with clinical manifestations differing with age, but most commonly include: diabetes insipidus, neurological and/or constitutional symptoms, and bone pain (described as nearly universal in presentation). Neurological involvement has been implicated as an independent predictor of death in ECD patients, occurring in 51% of the patients over the course of the disease and in 23% at disease onset. Diagnostic critera for this disease process are controversial and ever changing. Biopsy demonstrating characteristic histopathologic features in addition to clinico-radiographic features, most often sclerosing long bone involvement, is required to establish a diagnosis. The major criterion is the distinct histological pattern that characterizes the condition. Histological findings are sufficient to confirm ECD, which includes the presence of foamy histiocytosis with signs of inflammation Touton-type giant cells. Numerous laboratory and radiographic analyses are needed upon diagnosis to ascertain the extent of disease. Mutational analysis establishing BRAF and RAS mutational status is needed as well, as these bear implications for therapy with BRAF inhibition. Overall results of treatment have been generally disappointing as reported in one of the largest reviews on the disease by Veyssier-Belot et al., nearly 60% mortality within a 2.7 years follow up [2].
We describe a case report of an indolent form of Erdheim Chester Disease (ECD), in a patient in whom the disease has been present for approximately 14 years. Despite widespread sclerosis of long bones in the extremities, the patient has no complaints of peripheral bone pain. Although, the patient does have involvement of the jaw with subsequent loss of multiple teeth secondary to the facial manifestations of the disease. This is also the first patient have the disease incidentally discovered after an ACL injury, and the first ECD patient to receive an ACL reconstruction.
Case Report
Patient SF at the time of initial presentation was a 54 year old female, presenting to her family practice physician secondary to knee pain. She incurred a “knee sprain” while on vacation the week prior. Upon plain film imaging performed in the primary care provider’s office, marked boney sclerosis was noted. The patient was referred to an orthopedic surgeon for further evaluation. Orthopedic evaluation revealed a ruptured ACL, and the boney sclerosis was worrisome for an underlying pathological state. After appropriate radiological and laboratory workup by the orthopedic physician, she was referred to an oncologist for further evaluation. MRI imaging in combination with laboratory findings pointed toward myeloma. During the ACL Reconstruction, a boney biopsy was taken and subsequent biopsies in the future eventually revealed Erdheim Chester Disease. Immunochemistry noted positive for CD68, CD1a and S100 markers. The histology was characteristic of EDC, as explained previously. The histology differed from that of acute tendon injury. This typically includes fibroblasts surrounded by a collagen extracellular matrix, elastin, mucopolysaccharides, glycoproteins, and acute inflammatory cells. The patient over the course of 14 years has developed jaw pain and biopsies of the region revealed EDC in this region as well. The effects on the jaw have led to the loss of numerous teeth and jaw pain. The patient eventually developed acute renal failure, and exophthalmos, for which she underwent a decompressive procedure by an ophthalmologist. She is currently taking combinations of mediations to treat the EDC, which includes interferon therapy.
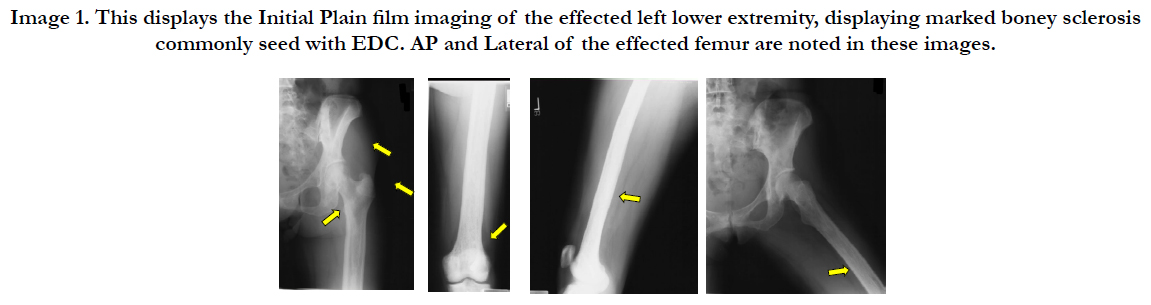
Image 1. This displays the Initial Plain film imaging of the effected left lower extremity, displaying marked boney sclerosis commonly seed with EDC. AP and Lateral of the effected femur are noted in these images.
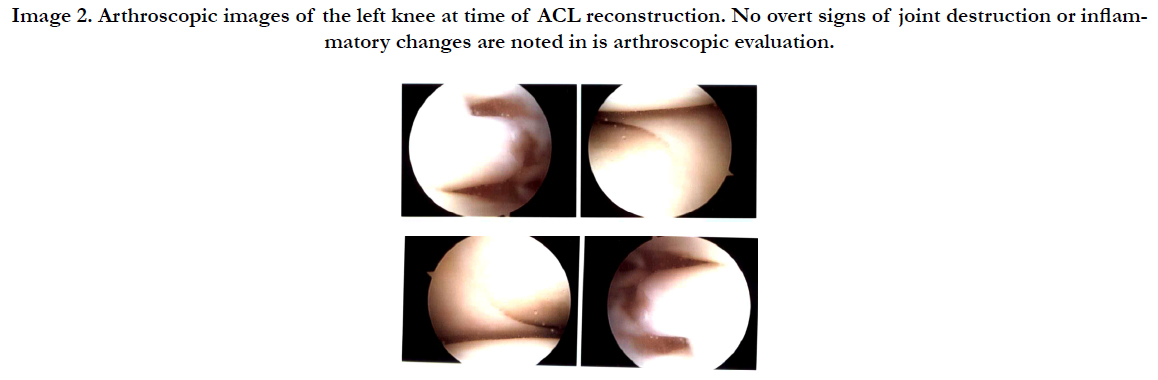
Image 2. Arthroscopic images of the left knee at time of ACL reconstruction. No overt signs of joint destruction or inflammatory changes are noted in is arthroscopic evaluation.
Discussion
The patient initially presented to physicians with pain at the knee secondary to a “sprain” she incurred while on vacation, which subsequently was diagnosed as an ACL rupture. This is the first case to present in this manner. Most patients present with a combination of bone pain, diabetes, and neurological symptoms, of which this patient had none. The patient underwent ACL reconstruction with bone-patellar-bone allograft without any complications. In a previous case report describing bilateral total knee arthroplasty in a patient with ECD, the patient was noted to have severe villous synovitis, as well as bone which was so dense/ sclerotic; “Because of the massive osteosclerosis of the trabecular bone and obliteration of the marrow space, it was necessary to place the intramedullary alignment guide ….for the distal cut of the femur in the correct position by drilling the femoral canal under fluoroscopic control. …..the osseous cuts required three saw blades for each cut as a result of the osteosclerosis of the distal part of the femur and proximal part of the tibia. Following ligament balancing of the extension gap, the final cuts of the femur with the saw guide required two additional saw blades” [3]. Although sclerosis was noted on pre-operative imaging, the bone was not so dense as to cause deviations in surgical procedure as seen in the case report of the total knee arthroplasty case. In fact, the ACL reconstruction occurred as normal performed, and there were no obvious signs of inflammatory changes intra-articularly as noted in the total knee case report with villous synovitis. It was previously speculated that severe synovitis and inflammatory markers intra-articularly could contribute the progression of cartilage destruction and joint disease as seen in previous case reports. The patient in this case report displayed nearly pristine joint space, with no obvious signs of synovitis or joint destruction (see Image 2). This paperdispels previous speculations ECD could have contributed to the synovitis, overall inflammatory environment in the knee, and overall joint destruction.
The sclerotic boney nature of the disease may lead to changes in how the ligamentous attachments of the bone absorb and transmit stress from the joint, secondary to abnormal interactions at the bone-ligament interface. We propose the idea that these sclerotic bones changes may predispose patients to ligamentous injuries as noted in this patient. Severe villous synovitis at the knee has been noted in patients with EDC by orthopedic surgeons in the past. Although no overt inflammatory changes were seen intra-articularly with the patient in question, microscopic changes may have been evident in the ligamentous structure of the ACL, which may have in itself lowered the threshold for incurring an injury. Although, these are both novel idea and more research is needed in the field to verify such speculations.
This case may represent an indolent form of the disease not previously revealed. Previous studies within the past 4-5 years have begun to investigate genetic linkages to the disease, but the genetic linkage is not clear. The patient in this case report has experienced renal disease and neurological symptoms (in the form of exophthalmos), but lacks many most common presentations seen in the disease (as previously described). She also has survived over 14 years with the pathology, with a disease mortality of around 60% in less than 3 years. Despite the independent risk factor of neurological symptoms as a risk factor for death, this patient continues to preserver. This is the 8th case in the medical literature to describe a patient with jaw involvement in the setting of ECD [4-10]. The addition of this rare entity of the disease, in combination with rare presentation as described thus far, makes this an exceedingly rare presentation. We propose the idea of variations in the genetic mutations, and call for more research to be done into the field to better diagnose patients and give them prognostic outlook on their condition.
References
- Cavalli G, Guglielmi B, Berti A, Campochiaro C, Sabbadini MG, et al., (2013) The multifaceted clinical presentations and manifestations of Erdheim- Chester disease: comprehensive review of the literature and of 10 new cases. Ann Rheum Dis. 72(10): 1691-5.
- Veyssier-belot C, Cacoub P, Caparros-lefebvre D, Brun B, Petit H, et al., (1996) Erdheim-Chester disease. Clinical and radiologic characteristics of 59 cases. Medicine (Baltimore). 75(3): 157-69.
- Steinert AF, Reppenhagen S, Baumann B, Rudert M, Nöth U (2011) Total knee arthroplasty in a patient with Erdheim-Chester disease with massive joint destruction: a case report. J Bone Joint Surg Am. 93(7): e29.
- Manaka K, Makita N, Iiri T (2014) Erdheim-Chester disease and pituitary involvement: a unique case and the literature. Endocr J. 61(2): 185-94.
- Nagatsuka H, Han PP, Taguchi K, Gunduz M, Nagai N, et al., (2005) Erdheim- Chester disease in a child presenting with multiple jaw lesions. J Oral Pathol Med. 34(7): 420-2.
- Kirchner TH, Seipelt G, Vogl TJ (2001) Involvement of the facial skull in Erdheim-Chester disease. Rontgenpraxis. 54(4): 148-51.
- Petrikowski CG, Mcgaw WT (2000) Erdheim-Chester disease of the jaws: literature review and case report. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 90(3): 389-98.
- Dinkar AD, Spadigam A, Sahai S (2007) Oral radiographic and clinicopathologic presentation of Erdheim-Chester disease: a case report. Oral Surg Oral Med Oral Pathol Oral RadiolEndod. 103(5): e79-85.
- Brahim JS, Guckes AD, Rudy SF (1992) Implant rehabilitation in Erdheim- Chester disease: a clinical report. J Prosthet Dent. 68(3): 399-401.
- Valdez IH, Katz RW, Travis WD (1990) Premature alveolar bone loss in Erdheim-Chester disease. Oral Surg Oral Med Oral Pathol. 70(3): 294-6.





