Treatment of Skin Aging and Photoaging with Innovative Oral Dosage Forms of Non-Hydrolized Carnosine and Carcinine
Babizhayev MA*
Innovative Vision Products, Inc., 3511 Silverside Road, Suite 105, County of New Castle, Delaware USA.
*Corresponding Author
Dr. Mark A. Babizhayev,
Innovative Vision Products, Inc.,
3511 Silverside Road, Suite 105, Country of New Castle, Delaware 19810, USA.
E-mail: markbabizhayev@yahoo.com
Received: May 31, 2017; Accepted : June 23, 2017; Published: June 30, 2017
Citation: Babizhayev MA (2017) Treatment of Skin Aging and Photoaging with Innovative Oral Dosage Forms of Non-Hydrolized Carnosine and Carcinine. Int J Clin Dermatol Res. 5(5), 116-143. doi: dx.doi.org/10.19070/2332-2977-1700031
Copyright: Babizhayev MA© 2017. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
Abstract
Aging is a multifactorial process resulting in several functional and esthetic changes in the skin. Advances in research have yielded a tremendous amount of information on the molecular pathways involved in both intrinsic aging (natural) and extrinsic aging (including photoaging). Some of the characteristic features of aging skin, such as wrinkling, loss of elasticity, and atrophy, can largely be attributed to dermal changes. The amount of collagen in the skin decreases, while the cross linking increases, and the solubility of collagen is reduced. The role of fibroblasts in aging tissue has been most extensively studied in mammalian skin. The total number of fibroblasts decreases, and their metabolism shows characteristic alterations.The dermis is maintained in large part by fibroblasts, which secrete dermal collagens, elastin, and other extracellular matrix components. When the skin is wounded, fibroblasts secrete proteases to degrade the wounded matrix, and then synthesize new matrix. The fibroblasts also secrete growth factors to stimulate the keratinocytes to proliferate and close the wound and cytokines to attract macrophages to engulf and degrade debris. Stress-induced premature senescence (SIPS) occurs after many different sublethal stresses including a number of oxidation factors, such as H2O2, hyperoxia, or organic and lipid hydroperoxides. Fibroblast cells in replicative senescence share common features with cells in SIPS: morphology, senescence-associated beta-galactosidase activity, cell cycle regulation, gene expression and telomere shortening. Most human cells lack sufficient telomerase to maintain telomeres, hence these genetic elements shorten with time and stress, contributing to aging and disease. We systematically examine the evidence supporting the use of dosage forms of non-hydrolized carnosine or carcinine in oral formulations for skin beautification purposes and provide a summary of the biomarkers of intrinsic and extrinsic skin aging, including photoaging. Senescence phenotype of human diploid fibroblasts is related with the exhaustion of their proliferative potential. This work suggests that different cell types, such as human skin fibroblasts, may use specific cellular treatment strategies with imidazole-containing dipeptides to halt the accelerated senescence of the fibroblast cells in response to telomere attrition and thus prevent skin aging through the number of biologically viable and safe metabolic pathways. The published data demonstrate that telomerase is expressed in the epidermis in situ independent of age. The reason for the sustained telomere length or expression of telomerase activity in the epidermis is associated not only with an increased turnover of keratinocytes, but also occurs due to the fact that the formation of a well structured epidermis strictly depends on a tight balance between proliferation and differentiation. Oral dosage forms of non-hydrolized carnosine or carcinine induce cellular responses in human skin fibroblasts through the telomere-mediated pathway and redox signaling, supporting the view that carnosine or related imidazole-containing dipeptide based compound-induced hormetic stimulation of cellular antioxidant defenses can be a useful approach toward anti-aging intervention to the skin.
2.Introduction
3.The Central Role of Fibroblast Cells Physiological Changes in Aging Skin Stroma
4.Variants, Causes and Factors of Skin Aging
5.Biomarkers of Intrinsic Skin Aging: Telomeres, Hormone Activity and Other Intrinsic Factors that Affect Manifestations of Skin Aging
6.Therapeutic Solutions to Improve Aging Skin
7.Conclusiong
8.Acknowledgements
9.References
Keywords
Skin Aging; Intrinsic Aging, Extrinsic Aging And Photoaging; Telomeres And Telomerase Biology; Epidermis, Skin Fibroblasts; Healthy Aging, Age-Related Diseases; Natural Imidazole-Containing Peptidomimetics; Oral Dosage Forms of Non-Hydrolized Carnosine or Carcinine; Cumulative Oxidative Stress; Smoking Therapeutic Nutritional Support; Telomere Length Protection; Skin Beautification.
Introduction
Aging is a multifactorial process resulting in several functional and esthetic changes in the skin. Recent advances in skin biology have increased our understanding of skin homeostasis and the aging process, as well as the mechanisms by which ultraviolet radiation contributes to photoaging and cutaneous disease. These advances in skin biology have led to the development of a diversity of treatments aimed at preventing aging and rejuvenating the skin [1]. The quest for youth and beauty of the skin is an ongoing one. In developed countries, interest in cutaneous aging is in large part the result of a progressive, dramatic rise over the past century in the absolute number and proportion of the population who are elderly. Young skin is characterized by thick epidermal and dermal layers and relatively efficient wound healing. The epidermis contains numerous invaginations, or rete ridges, and the dermis contains dense collagen.
Old skin, by contrast, is characterized by a thinner epidermis that contains fewer and shallower rete ridges. The dermis also becomes thinner, showing a marked loss of collagen and other fibers. Changes in the dermis are in large measure responsible for the loss of elasticity and wrinkling that is the hallmark of aging skin. In addition, wound healing slows with age. Aging is particularly sensitive to environmental influence in the skin: skin exposed to the sun (ultraviolet light) ages much more rapidly than sun-protected skin.
The aging and its effects on the skin in older persons have created a demand for a better understanding of the aging process and particularly for effective therapeutic interventions for better skin beautification and rejuvenation. Skin aging and longevity regulation are complex processes determined by the genetic endowment of the individual and by environmental factors.
The appearance of old skin and the clinical consequences of skin aging have been well known for centuries, but it is only in the past 50 years that mechanisms and mediators have been pursued systematically. Still, within a relatively short time, there has been tremendous progress, a progress greatly enhanced by basic gerontologic research using immunologic, biochemical, cellular and in particular, molecular biologic techniques [2]. An impressive amount of basic science and clinical research has been conducted in both an attempt to discover novel strategies for preventing detrimental sun damage and to validate the addition of novel oral antioxidant formulations to skin care products. As dermatologists, it will be essential to provide our patients with substantiated counseling regarding the efficacy of commercial assertions. The average age of people has been increasing over the years, triggering more awareness and more interest in the study of regenerative medicine, especially degeneration of the skin which is an organ that is crucial noticeably for appearance.
In this review, we will systematically examine the evidence supporting the use of patented dosage forms of non-hydrolized carnosine or carcinine in oral formulations for skin beautification purposes and provide a summary of the pathogenesis of intrinsic and extrinsic skin aging , involved factors of aging and photoaging. This review article suggests that different cell types , such as human skin fibroblasts may use specific cellular treatment strategies with imidazole-containing dipeptides to halt the accelerated senescence of the fibroblast cells in response to telomere attrition and thus prevent skin aging through the number of metabolic pathways. This may be attributed to the distinct senescent phenotypic characteristics of the resident skin fibroblasts. The results suggest that ectopic expression of hTERT, in addition to acting in telomere length maintenance by safe activating telomerase with imidazole-containing dipeptide(s), also functions in regulating senescence induction.
The Central Role of Fibroblast Cells Physiological Changes in Aging Skin Stroma
Skin aging is the multifactorial process both internal and external factors, such as, age, sex, race, disease of internal organs and environmental exposure. However, the main causes of skin degeneration are heredity and sunlight. The latter induces the most skin degeneration. Thus far, many studies have been conducted on pathogenesis and prevention of Photo-aging, as well as regeneration of damaged skin [3-8]. The role of fibroblasts in aging tissue has been most extensively studied in mammalian skin. The skin is composed of two primary layers: the epidermis, which contains the major epithelial cells of the skin (epidermal keratinocytes), and the dermis, which is the stromal layer of the skin (Figure 1, Tables 1, 2). As with other stroma, the dermis is maintained in large part by fibroblasts, which secrete dermal collagens, elastin, and other extracellular matrix components. When the skin is wounded, fibroblasts secrete proteases to degrade the wounded matrix, and then synthesize new matrix. The fibroblasts also secrete growth factors to stimulate the keratinocytes to proliferate and close the wound and cytokines to attract macrophages to engulf and degrade debris [9].
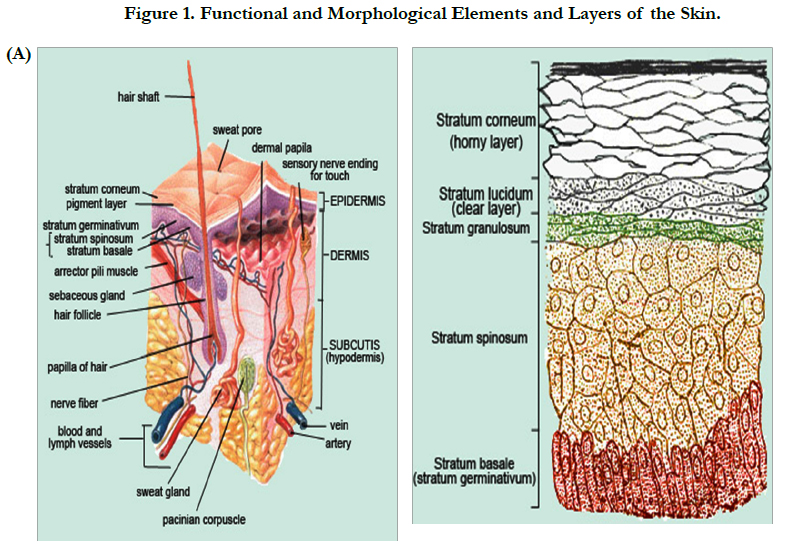
Figure 1. Functional and Morphological Elements and Layers of the Skin.

Above Figure Description:
A. The stratum corneum ("the horny layer") is the outermost layer of the epidermis (the outermost layer of the skin). It is composed mainly of dead cells that lack nuclei. As these dead cells slough off, they are continuously replaced by new cells from the stratum germinativum (basale). In the human forearm, for example, about 1300 cells/cm2/hr are shed and commonly accumulate as house dust. Cells of the stratum corneum contain keratin, a protein that helps keep the skin hydrated by preventing moisture evaporation. In addition, these cells can also absorb moisture, further aiding in hydration and explaining why humans and other animals experience wrinkling of the skin. Keratinocyte is the major cell type of the epidermis, making up about 90% of epidermal cells. The epidermis is divided into four or five layers (depending on skin type) based on keratinocyte morphology:
• Stratum basale (at the junction with the dermis)
• Stratum spinosum
• Stratum granulosum
• Stratum lucidum (only present in thick skin - i.e. palms of hand and soles of feet)
• Stratum corneum
Keratinocytes originate in the basal layer from the division of keratinocyte stem cells. They are pushed up through the layers of the epidermis, undergoing gradual differentiation until they reach the stratum corneum where they form a layer of enucleated, flattened, highly keratinized cells called squamous cells. This layer forms an effective barrier to the entry of foreign matter and infectious agents into the body and minimises moisture loss.
Taking non-hydrolized imidazole-containing dipeptide based antioxidant oral supplements can help enhance the skin’s natural protective properties (see below) as well as having beautifying properties.
Skin is composed of the dermis and epidermis - each with unique components and functions. Contained within each layer are mixtures of cells and connective tissue that provide form and function for the skin (Figure 1). More specifically, keratinocytes, which produce keratin to harden and waterproof the skin, can be found in the epidermal layer. Melanocytes, which produce melanin, provide pigmentation for both hair and skin, deliver protection from UV radiation, and are found in fewer numbers in the dermal layer. Melanocytes intercalate up into the epidermis and establish close and critical interactions with keratinocytes to perform various cellular functions during development and normal maintenance of the skin. Healthy skin also contains a number of other structural elements, including collagen fibers and fibroblasts located in the basement membrane, lending strength and structure to the skin. Layered within the basement membrane matrix are dermal microvasculature and lymphatic vessels for blood circulation and waste removal, vital to proper skin function.
Normal skin is shown with the key epidermal and dermal elements. The epidermis contains the dividing epithelial cells at the basement membrane migrating to the surface forming keratinocytes and maintaining the surface barrier. The basement membrane, rich in adherence proteins, anchors the epithelial cell layer. The dermis is made of components produced by fibroblasts, including collagen (mostly type I), glycosaminoglycans, and other matrix proteins.
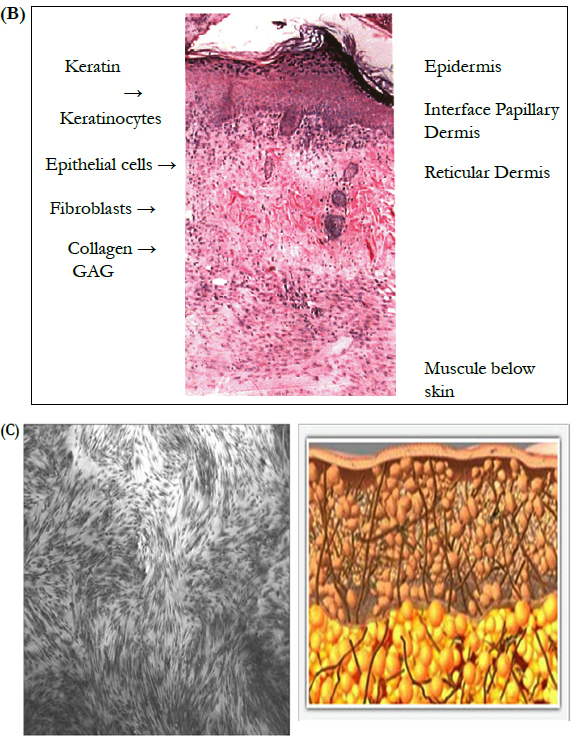
B. The cross section of normal skin demonstrates the key components. Keratin, a product of keratinocytes, resides on the outer most surface. Keratinocytes (epithelial cells) make up the first or outer layer of skin, i.e. the epidermis. Note that keratinocytes can also be seen in the cross sections of hair follicles found in the dermis. The dermal layer is composed of collagen seen as swirls of pink material. The lighter pink homogenous material is made up of glycosaminoglycans and other compounds produced by the fibroblasts (elongated dark cells) such as fibronectin and hyaluronic acid.
Dermis:
Protection from trauma due to the dermal properties of elasticity and durability.
Fluid balance through regulation of skin blood flow.
Thermoregulation through control of skin blood flow.
Growth factors and contact direction for epidermal replication and dermal repair.
C. Healthy human fibroblasts
Simultaneously dermal activity stimulates the skin’s fibroblasts to increase collagen replenishment to strengthen and rejuvenate skin for a smoother, tighter, more contoured appearance.


Table 1. Characteristics of the Skin Layers.

Table 2. Skin Functions and Characteristics of cell Elements of the Skin.
The life history of fibroblast and fibroblast-like cells includes an initial stage of outgrowth and establishment in culture; a period of vigorous proliferation which has a variable length, depending on the tissue of origin, age of the donor, etc.; a period of declining proliferative vigor which includes substantial cell death; and finally, the emergence of an (apparently) long-lived population which is unable to proliferate in response to growth factors. The research in this area began with a detailed characterization and comparison of young versus senescent cell morphology and physiology. These studies provided the basis for a wide variety of subsequent studies that addressed possible mechanisms underlying cell senescence. These included studies on DNA repair, protein synthetic errors, chromatin structure and function, and mechanisms for modulating replicative life span (reviewed in ref. [10]). Senescent fibroblasts appear to increase with age in human dermis, and senescent fibroblasts constitutively secrete factors that, ordinarily, are secreted only transiently during wound healing [10, 11]. These factors include interstitial collagenase and elastase, which are matrix metalloproteinases that degrade dermal collagens and elastin. Ultraviolet light can also induce these metalloproteinases, as well as cellular senescence, in fibroblasts. Thus, some of the hallmarks of aging skin, such as wrinking and loss of dermal elasticity, are likely due, at least in part, to the secretion of metalloproteinases by fibroblasts, which, in turn, may be due to cellular senescence and/or environmental exposure to ultraviolet light.
A major factor that determines the appearance of the skin is the condition of the dermal collagen. Although there are differences between young and aged skin with respect to the condition of the epidermis, dermal collagen is the most important factor in whether the skin appears to be young or old [12]. Some of the characteristic features of aging skin, such as wrinkling, loss of elasticity, and atrophy, can largely be attributed to dermal changes. The amount of collagen in the skin decreases, while the cross linking increases, and the solubility of collagen is reduced. The total number of fibroblasts decreases, and their metabolism shows characteristic alterations. Some of the functions of fibroblasts in aging skin, such as the synthesis of protein and collagen, but also proliferation and chemotaxis, can be investigated by means of in vitro models. In addition, various syndromes of premature aging (e.g. progeria, Werner's syndrome) have proved useful models and have contributed considerably to the understanding of aging processes [13]. On a molecular level, UV radiation from the sun attacks keratinocytes and fibroblasts, resulting in the activation of cell surface receptors, which initiate signal transduction cascades. This in turn leads to a variety of molecular changes, which causes a breakdown of collagen in the extracellular matrix and a shutdown of new collagen synthesis [14]. Research on fibroblast biology in three-dimensional collagen matrices offers new opportunities to understand the reciprocal and adaptive interactions that occur between cells and surrounding matrix in a tissue-like environment. Such interactions are integral to the regulation of connective tissue morphogenesis and dynamics that characterizes tissue homeostasis and wound repair. During fibroblast collagen matrix remodeling, mechanical signals from the remodeled matrix feed back to modulate cell behavior in an iterative process. As mechanical loading (tension) within the matrix increases, the mechanisms used by cells to remodel the matrix change. Fibroblasts in matrices that are under tension or relaxed respond differently to growth factor stimulation, and switching between mechanically loaded and unloaded conditions influences whether cells acquire proliferative/biosynthetic active or quiescent/resting phenotypes [15].
Another feature of aged skin is fragmentation of the dermal collagen matrix. Fragmentation results from actions of specific enzymes (matrix metalloproteinases) and impairs the structural integrity of the dermis. Several of metalloproteinases, as for instance MMP-2 and MMP-9 among others also act on elastic fibers (reviewed in ref. [16]). Fibroblasts that produce and organize the collagen matrix cannot attach to fragmented collagen. Loss of attachment prevents fibroblasts from receiving mechanical information from their support, and they collapse. Stretch is critical for normal balanced production of collagen and collagen-degrading enzymes. In aged skin, collapsed fibroblasts produce low levels of collagen and high levels of collagen-degrading enzymes. This imbalance advances the aging process in a self-perpetuating, neverending deleterious cycle. Clinically proven anti-aging treatments such as topical retinoic acid, carbon dioxide laser resurfacing, and intradermal injection of cross-linked hyaluronic acid stimulate production of new, undamaged collagen. Attachment of fibroblasts to this new collagen allows stretch, which in turn balances collagen production and degradation and thereby slows the aging process. Collagen fragmentation is responsible for loss of structural integrity and impairment of fibroblast function in aged human skin. Treatments that stimulate production of new, nonfragmented collagen should provide substantial improvement to the appearance and health of aged skin (reviewed in ref. [16]).
The dermal extracellular matrix among the other molecular constituents is composed of type I collagen, proteoglycans, and glycosaminoglycans produced and secreted by fibroblasts. Type I collagen is the most abundant protein in the dermal extracellular matrix. It is responsible for the support and structure of the skin, and its loss and degradation are thought to result in the clinical phenotype of aged skin, specifically with regard to fine and coarse wrinkles [12-18]. Elastic fibers are another key protein in skin and reside in the papillary and reticular dermis. They provide elasticity and recoil capacity to skin. Certain diseases of elastic fibers, such as Marfan’s syndrome (fibrillin-1 mutation) and Williams syndrome (elastin mutations), are characterized by loss of elasticity, much like that seen in aged skin. Clearly, loss and damage of elastic fibers are contributory to the clinical and molecular changes seen in aged skin, but, for practical reasons, the majority of the research work being carried out is focused on collagen [12].
Collagen fibers provide “scaffolding” for fibroblasts; this scaffolding is critical for the maintenance of the collagen network because it allows the fibroblasts to exist in a “stretched” configuration, as a typical of young, healthy, non-sun-damaged skin [15, 16]. The fibroblasts appear elongated, with abundant endoplasmic reticulum indicative of the active collagen protein-producing machinery. Collagen fibrils are intimately associated with these elongated fibroblasts and are seen in close proximity to them. In contrast, the fibroblasts in old and sun-damaged skin exist in a “collapsed” configuration, and the cells appear much smaller than the stretched fibroblasts [12,15,16]. There is minimal endoplasmic reticulum in these fibroblasts as seen by electron micrography, indicating that the collagen producing machinery is minimally productive. In old, sun-damaged skin, collagen fibrils are not seen in close proximity external to the fibroblasts and are fragmented. Therefore, it is important that the relationship between the fibroblasts and the collagen matrix is an interdependent one, with the fibroblasts serving as the structure or anchor for the collagen fibers and the collagen fibers allowing the fibroblasts to exist in their “stretched” configuration [12,15,16].
The results demonstrate that age-related alterations in collagen and total protein metabolism of skin fibroblasts in culture were similar to those reported previously for skin in vivo, suggesting that for studies of these processes, fibroblasts in culture provide an appropriate model [19]. Age-related changes in collagen synthesis in rat skin fibroblasts in vitro over 30 population doublings were determined based on the production of hydroxy-[14C]pro-line. Degradation of newly synthesized collagen was based on the appearance of free hydroxy-[14C]proline in the culture system [19]. Total protein synthesis rates were based on the incorporation of [14C]proline into proteins. in vitro rates of collagen synthesis decreased 5-fold over 30 population doublings (P < 0.05). Degradation of newly synthesized collagen increased from 33.0 +/- 0.8% (n = 4, SEM) to 45.2 +/- 1.1% (n = 4; P < 0.05) over the same period, with a maximum after 25 population doublings of 55.8 +/- 1.1% (n = 4). Total protein synthesis rates decreased by one-half over 30 population doublings (P < 0.05). The results indicated that collagen production in rat decreased as cells aged in vitro and that this was due to both changes in synthesis and degradation. Senescent and ultraviolet-damaged fibroblasts also secrete enzymes that degrade the basement membrane, the dense matrix onto which the epithelial cells are organized. This may contribute to the age-dependent thinning of the epidermis and the loss of rete ridges, as the basement membrane is critically important for the proper organization and function of epithelial cells [19]. Among the factors involved, the accumulation of advanced glycation end-products (AGEs) might well play an important role. Several of such AGE-products showed a significant inhibition of collagen deposition. On the contrary, retinol, ascorbic acid as well as the rhamnose-rich oligo-and polysaccharides (RROPs) did produce a significant upregulation collagen deposition. Polysaccharide preparations, rich in rhamnose and fucose (the EROBmixture) could protect against the AGEs-induced inhibition of collagen accumulation [20].
Extrinsic aging is associated with episodic photodamage, which culminates in permanent photoaging. Minimal erythema dose refers to the amount of UV radiation that causes barely perceptible skin reddening. A mere 0.01 minimal erythema dose leads to statistically significant injury that will induce the molecular sequence of events resulting in collagen deficiency, collagenase excess, and micro- and macro-scarring leading to solar scars. These additive solar scars are associated with the clinical presentation of wrinkles [12]. Both UVB and UVA radiation lead to an increase in reactive oxygen species (ROS). Specifically, exposure to two minimal erythema doses leads to the production of H2O2, which gives rise to other oxygen free radicals within minutes. ROS play an important role in direct cellular damage (cell walls, lipid membranes, mitochondria, and DNA) as well as in molecular signaling. Through ROS signaling, transforming growth factor-, a cytokine that promotes collagen production, is blocked, and the formation of new collagen is reduced. Also mediated through ROS signaling is the epidermal growth factor receptor pathway, which signals activator protein-1, a transcription factor responsible for upregulating collagenase, a matrix metalloproteinase that is important in collagen breakdown. The upregulation of collagenase leads to an increase in collagen fragmentation that in turn leads to a decrease in the mechanical tension of the fibroblast [12]. Loss of mechanical tension leads to perpetuation of the cycle in which collagenase concentration is increased, leading to further collagen fragmentation and, ultimately, permanent collagen loss clinically manifested as permanent photoaging. Each UV insult results in solar scars, which manifest as a wrinkle [21,22]. Long-term exposure to ultraviolet irradiation from sunlight causes premature skin aging (photoaging), characterized in part by wrinkles, altered pigmentation, and loss of skin tone. Multiple exposures to ultraviolet irradiation lead to sustained elevations of matrix metalloproteinases that degrade skin collagen and may contribute to photoaging [22].
Photoaged skin displays prominent alterations in the collagenous extracellular matrix of connective tissue. Molecular mechanisms by which ultraviolet light causes photoaging involve activation of growth factor and cytokine receptors in keratinocytes and dermal cells. They lead to downstream signal transduction through activation of mitogen-activated protein kinase (extracellular signal-regulated kinase, c-jun N-terminal protein kinase, and p38) pathways. These signaling pathways converge in the nucleus of cells to form an activated complex of transcription factor activator protein 1 (cFos/cJun), which induces matrix metalloproteinases that degrade skin connective tissue. In addition to cell surface receptor activation, generation of ROS by ultraviolet radiation is believed to be critical in triggering mitogen-activated protein kinase pathways. Kang et al., [21] investigated the ability of (i) ultraviolet irradiation to generate ROS in human skin in vivo; and (ii) genistein, which possesses both tyrosine kinase inhibitory and antioxidant activities, and n-acetyl cysteine, which can be converted into the endogenous antioxidant glutathione, to impair responses to ultraviolet light that eventuate in photoaging in human skin in vivo. Ultraviolet irradiation caused a rapid and significant increase in hydrogen peroxide levels in human skin in vivo. Pretreatment of human skin with genistein inhibited ultraviolet-induced epidermal growth factor receptor tyrosine kinase activity, whereas n-acetyl cysteine did not. Genistein inhibited ultraviolet induction of both extracellular signal-regulated kinase and cJun N-terminal protein kinase activities. n-Acetyl cysteine inhibited extracellular signalregulated kinase but not cJun N-terminal protein kinase activation. Both genistein and n-acetyl cysteine prevented ultraviolet induction of cJun protein. Consistent with this, genistein and n-acetyl cysteine blocked ultraviolet induction of cJun-driven enzyme, collagenase. Neither genistein nor n-acetyl cysteine acted as sunscreens as they had no effect on ultraviolet-induced erythema. These data indicate that compounds similar to genistein and nacetyl cysteine, which possess tyrosine kinase inhibitory and/ or antioxidant activities, may prevent photoaging [21]. N-acetylcysteine has been used as the active ingredient in the formulation of non-hydrolized carnosine proposed in this study (Figure 7).
The sequence of events in intrinsic aging is similar but with a few notable exceptions. The damage in natural aging is continuous rather than episodic and is caused by the passage of time rather than by UV damage. The increase in ROS signaling leads to a decrease in collagen formation mediated by transforming growth factor-, as in extrinsic aging. The increase in collagenase level, however, is mediated through the JNK pathway rather than through the epidermal growth factor receptor pathway. The above discussion is focused on collagen in aging skin because most of the ongoing research is focused on this vital skin protein. There have, however, been findings to suggest that there is a difference between photoprotected and photodamaged skin with regard to extracellular protein matrix. The extracellular matrix is important because of its water-binding capacity. Four glycosaminoglycans were identified in skin samples: hyaluronic acid (HA), heparan sulfate, dermatan sulfate and chondroitin sulfate [12,23]. In a study of photodamaged facial skin as compared with photoprotected postauricular skin used as a control, photoexposed skin was observed to have a significant increase in the content of HA and DS as compared with photoprotected skin. The increase in HA was associated with an increase in the amount of fragmented HA, which is abnormal and does not interact in the usual manner with water [12,24].
Age-dependent changes in fibroblast physiology (senescent phenotype of fibroblasts) may also contribute to the increased incidence of cancer that is a hallmark of mammalian aging. Several lines of evidence suggest mutations and loss of normal tissue structure synergize to generate the exponential rise in cancer that occurs with age [25]. Tissue structure and integrity are critically dependent on an intact stromal and basement membrane, both of which are disrupted by senescent or damaged fibroblasts. In addition, senescent fibroblasts secrete epithelial growth factors, which can stimulate the growth of epithelial cells that have acquired potentially oncogenic mutations.
In summary of this section of the article, fibroblasts undergo physiological changes with age. These changes are induced by environmental and intrinsic factors, and disrupt the integrity of the stroma and basement membrane. Both these structures are critical in order for epithelial cells, and hence tissues, to carry out their normal functions. These structures are also important for suppressing the progression of cancer.
Variants, Causes and Factors of Skin Aging
Advances in research have yielded a tremendous amount of information on the molecular pathways involved in both intrinsic aging (natural) and extrinsic aging (photoaging) [9]. Both genetic (intrinsic) and environmental (extrinsic) factors contribute to the phenotypic changes in cutaneous aging. Apart from them, so called stochastic aging connotes cell damage caused by metabolic processes, free radicals and cosmic irradiation. Extrinsically aged skin shows signs of photodamage which include appearance of wrinkles (Figure 2a), pigmented lesions (Figure 2b), actinic keratoses and patchy hypopigmentations. Extrinsic aging is caused by environmental factors such as sun exposure, air pollution, smoking, alcohol abuse, and poor nutrition.
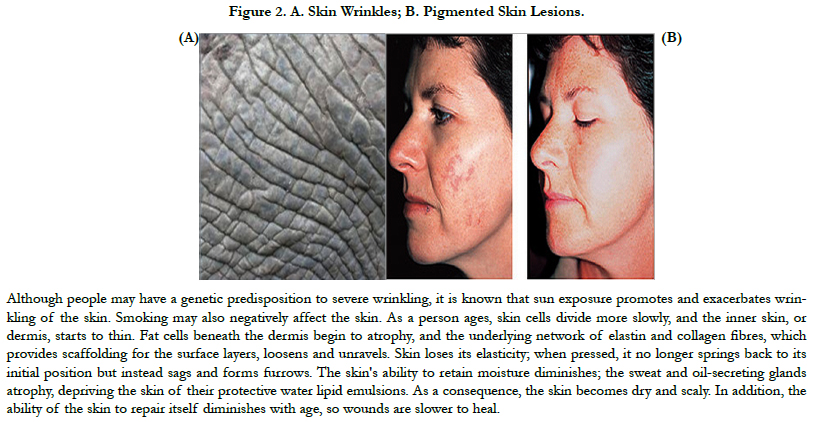
Figure 2. A. Skin Wrinkles; B. Pigmented Skin Lesions.
Intrinsic aging reflects the genetic background and depends on time. Various expressions of intrinsic aging include smooth, thinning skin with exaggerated expression lines. It is inevitable and time dependent.
Extrinsically aged skin is characterized by photo damage as wrinkles, pigmented lesions, patchy hypopigmentations, and actinic keratoses. Therapeutic modalities imply photoprotection with sunscreens that prevent sunburns and block ultraviolet irradiation.
Other modalities include use of retinoids which regulate gene transcription with subsequent cellular differentiation and proliferation. The topical and peroral administration of network antioxidants, such as vitamin E and C, coenzyme Q10, alpha-lipoic acid and glutathione, enhance anti-aging effect. The other antioxidants such as green tea, dehydroepiandrosterone, melatonin, selenium and resveratrol, have also antiaging and anti-inflammatory effects. Topical bleaching agents such as hydroquinone, kojic acid and azelaic acid can reduce signs of aging. Studies confirm the efficacy of these topical agents in combination with superficial and/or medium depth or deep peeling agents for photodamaged skin treatment. Indications for type of chemical peels according to various clinical diagnosis are done, as well as advantages and disadvantages of different types of chemical peels [3,26].
However, only recently have the underlying molecular mechanisms involved in these changes been elucidated. DNA damage to both genomic and mitochondrial DNA and subsequent DNA repair contribute greatly to age-associated skin changes and carcinogenesis. Better understanding of these intricate, interwoven mechanisms involved in DNA damage and repair might help to develop new strategies in preventing and treating changes of intrinsic skin aging and photoaging, improving skin appearance [27].
The dramatic increase in the aging population and the psychosocial impact of skin aging has created a demand for effective interventions. The advances that have been made in the past 25 years in our understanding of the clinical, biochemical, and molecular changes associated with aging have led to the development of many different approaches to reduce, postpone, and in some cases, repair the untoward effects of intrinsic programmed aging and extrinsic environmental injury [4, 27]. The pathogenesis of skin degeneration includes natural and aggravating factors [1,13- 15]. These factors are heredity which is internal and hard to control and environmental which is external [1,3,4-7,26]. Instances of the latter are cigarette smoke, water and air pollutions, and sunlight. Sunlight has the most influence on skin degeneration [3, 27]. Chronic sun exposure causes photoaging of human skin, a process that is characterized by clinical, histological and biochemical changes which differ from alterations in chronologically aged but sun-protected skin. Within recent years, substantial progress has been made in unraveling the underlying mechanisms ofphotoaging. Induction of matrix metalloproteinases as a consequence of activator protein (AP)-1 and nuclear factor (NF)-KB activation as well as mutations of mitochondrial DNA have been identified recently. This has increased our understanding of photoaging significantly and has led to new prophylactic and therapeutic strategies aimed at the prevention and repair of the detrimental effects of chronic sun exposure on the skin [8].
Severe oxidative stress progressively leads to cell dysfunction and ultimately cell death. Oxidative stress is defined as an imbalance between pro-oxidants and/or free radicals on the one hand, and anti-oxidizing systems on the other. The oxygen required for living may indirectly be responsible for negative effects; these deleterious effects are due to the production of free radicals, which are toxic for the cells (superoxide anion radicals, hydroxyl radicals, peroxyl radicals, hydrogen peroxide, hydroperoxides and peroxinitrite anions). Free radical attacks are responsible for cell damage and the targeted cells are represented by the cell membranes, which are particularly rich in unsaturated fatty acids, sensitive to oxidation reactions; DNA is also the target of severe attacks by these ROS [29,30].
These are represented by the enzymes and free radical captors. The latter are readily oxidizable composites. The free radical captor or neutralization systems of these ROS use a collection of mechanisms, vitamins (E and C), enzymes [superoxide dismutase (SOD), glutathione peroxidase (GPx) and others], and glutathion reductase (GSH), capable of neutralizing peroxinitrite. The efficacy of this system is dependent on the genome for the enzymatic defence systems, and on nutrition for the vitamins. Some strategies aimed at reducing oxidative stress-related alterations have been performed in animals. However, only a few can be used and are efficient in humans, such as avoidance of unfavourable environmental conditions (radiation, dietary carcinogens, smoking etc.) and antioxidant dietary supplementation.
Epidemiological data suggest that antioxidants may have a beneficial effect on many age-related diseases: atherosclerosis, cancer, skin appearance, some neurodegenerative and ocular diseases. However, the widespread use of supplements is hampered by several factors: the lack of prospective and controlled studies; insufficient knowledge on the pro-oxidant, oxidant and antioxidant properties of the various supplements; growing evidence that free radicals are not only by-products, but also play an important role in cell signal transduction, apoptosis and infection control [28,29].
Biomarkers of Intrinsic Skin Aging: Telomeres, Hormone Activity and Other Intrinsic Factors that Affect Manifestations of Skin Aging
Intrinsic aging, also known as the natural aging process, is a continuous process that normally begins in our mid-20s. Within the skin, collagen production slows, and elastin, the substance that enables skin to snap back into place, has a bit less spring. Dead skin cells do not shed as quickly and turnover of new skin cells may decrease slightly. While these changes usually begin in our 20s, the signs of intrinsic aging are typically not visible for decades. The signs of intrinsic aging are presented in Table 3. Genes control how quickly the normal aging process unfolds. Some notice those first gray hairs in their 20s; others do not see graying until their 40s. People with Werner’s syndrome, a rare inherited condition that rapidly accelerates the normal aging process, usually appear elderly in their 30s. Their hair can gray and thin considerably in their teens. Cataracts may appear in their 20s. The average life expectancy for people with Werner’s syndrome is 46 years of age.

Table 3. Clinical Biomarkers of Intrinsic Aging.
Intrinsic aging depends on time. The changes occur partially as the result of cumulative endogenous damage due to the continuous formation of ROS, which are generated by oxidative cellular metabolism. Despite a strong antioxidant defense system, damage generated by ROS affects cellular constituents such as membranes, enzymes, and DNA [26, 30,31]. It has a genetic background, but is also due to decreased sex hormone levels.
Telomeres are located at the end of the chromosomes and consist of an evolutionary conserved repetitive nucleotide sequence ranging in length from a few hundred base pairs in yeast till several kilo base pairs in vertebrates. The telomere, a terminal portion of the eukaryotic chromosome, plays an important role. With each cell division, the length of the human telomere shortens. In all mammals telomeres are short sequences of bases and are arranged in the same mode (TTAGGG) and so the enzyme telomerase is responsible for its maintenance (Figure 3) [32- 34]. Telomere protects the end of the chromosome from fusion or deletion and maintains the stability of the chromosome and is synthesized by telomerase, a ribonucleoprotein. Telomere reduction is observed with cell senescence and immortalization, both in vivo and in vitro. Thus, telomere is considered to be a "clock" which measures the life span of cells, and its length is altered by cellular senescence and immortalization [32]. Telomeres have been postulated as a universal biological clock that shortens in parallel with aging in cells [33, 34]. Even in fibroblasts of quiescent skin more than 30% of the telomere length is shortened during adulthood [33]. Telomeres associate with shelter in proteins and form a complex protecting the chromosomal deoxyribonucleic acid (DNA) from recognition by the DNA damage repair system. Due to the "endreplication problem" telomeres shorten with each mitotic cycle resulting in cumulative telomere attrition during aging. When telomeres reach a critical length the cell will not further undergo cell divisions and become senescent or otherwise dysfunctional. Telomere shortening has not only been linked to aging but also to several age associated diseases, including tumorigenesis, coronary artery disease, and heart failure [34]. In the current review, we discuss the role of telomere biology in relation to skin aging, senescence phenotype of the dermal fibroblasts and skin aging associated diseases.
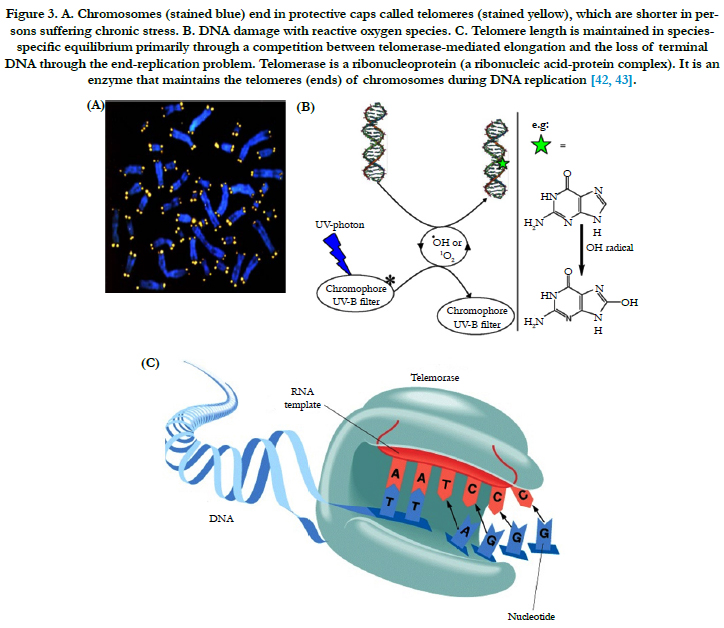
Figure 3. A. Chromosomes (stained blue) end in protective caps called telomeres (stained yellow), which are shorter in persons suffering chronic stress. B. DNA damage with reactive oxygen species. C. Telomere length is maintained in speciesspecific equilibrium primarily through a competition between telomerase-mediated elongation and the loss of terminal DNA through the end-replication problem. Telomerase is a ribonucleoprotein (a ribonucleic acid-protein complex). It is an enzyme that maintains the telomeres (ends) of chromosomes during DNA replication [42, 43].
Telomerase and telomeres have been shown to be involved in the control of cell proliferation, the regulation of cell senescence and the unlimited proliferation capacity of malignant cells (reviewed in ref. [35]). Human telomeres function to protect chromosomes from degradation, fusion and recombination. Since the termini of linear molecules are replicated only in the 5'-3' direction by conventional DNA polymerases and require an RNA primer to initiate DNA synthesis, the removal of the RNA primer results in DNA loss with each cell division. To date, telomere shortening has been observed in most dividing somatic cells, eventually leading to cell senescence when critically short telomeres are reached. Borderline telomerase activity has been detected in human primitive hematopoietic cells and in stimulated lymphocytes which increased with cytokine induced ex vivo expansion. However, in most other normal somatic cells, telomerase has not been detected, and consequently telomere shortening can be anticipated after a limited number of population doublings. In contrast, spontaneously immortalized tumor cell lines and the majority of malignant tumors demonstrate high telomerase activity, stable telomere length and unlimited proliferative potential. Mechanisms for telomerase and telomere length regulation are under extensive investigation. These have included the cloning of the RNA component and telomerase associated proteins, antisense experiments that have demonstrated progressive telomere length shortening in the absence of telomerase, and the identification of telomere binding proteins which may regulate telomerase by creating a negative feedback signal (reviewed in ref. [35]). Telomeres play a central role in cell fate and aging by adjusting the cellular response to stress and growth stimulation on the basis of previous cell divisions and DNA damage. At least a few hundred nucleotides of telomere repeats must "cap" each chromosome end to avoid activation of DNA repair pathways. Repair of critically short or "uncapped" telomeres by telomerase or recombination is limited in most somatic cells and apoptosis or cellular senescence is triggered when too many "uncapped" telomeres accumulate. The chance of the latter increases as the average telomere length decreases. The average telomere length is set and maintained in cells of the germline which typically express high levels of telomerase. In somatic cells, telomere length is very heterogeneous but typically declines with age, posing a barrier to tumor growth but also contributing to loss of cells with age [36].
Initially, when human fibroblasts from different donors are grown in vitro, only a small fraction of the variation in their finite replicative capacity is explained by the chronological age of the donor [33]. Because telomeres, the terminal guanine-rich sequences of chromosomes, shorten throughout the life span of cultured cells, Allsopp et al., [33] wished to determine whether variation in initial telomere length would account for the unexplained variation in replicative capacity. Analysis of cells from 31 donors (aged 0-93 yr) indicated relatively weak correlations between proliferative ability and donor age (m = -0.2 doubling per yr; r = -0.42; P = 0.02) and between telomeric DNA and donor age (m = -15 base pairs per yr; r = -0.43; P = 0.02). However, there was a striking correlation, valid over the entire age range of the donors, between replicative capacity and initial telomere length (m = 10 doublings per kilobase pair; r = 0.76; P = 0.004), indicating that cell strains with shorter telomeres underwent significantly fewer doublings than those with longer telomeres. These observations suggest that telomere length is a biomarker of somatic cell aging in humans and are consistent with a causal role for telomere loss in this process (Figure 4)[33]. Moreover, variation in the initial length of terminal restriction fragments (TRF) accounts for much of the variation in replicative capacity of fibroblast cultures from different donors. Since replicative capacity also varies significantly between clones in a mass culture of fibroblasts from a single donor, Allsopp et al., [37] wished to further test the hypothesis that the shortening of telomeres to a critical or threshold length acts as a signal for cell senescence. Thus, Allsopp et al., [37]] measured TRF length and total telomeric signal intensity for 35 clonal fibroblast populations at early passage and at senescence. Replicative capacity was found to be directly proportional to mean TRF length (m = 7.2 population doublings/kbp, r = 0.65, P = 0.0004) and total signal intensity (m = 25.0 population doublings/unit, r= 0.63, P < 0.003) at early passage. More importantly, the variability in both mean TRF length and signal intensity (F = 2.0 and 2.9; P = 0.02 and 0.03, respectively) at senescence was markedly less than that at early passage. Although initial telomere length cannot account for all of the interclonal variability in replicative capacity, the observations support the existence of a critical telomere length in senescing cells and a causal role of telomere shortening in cell senescence [37]. Thus, it has been documented that the loss of telomere repeats was causally linked to in vitro replicative senescence of human diploid fibroblasts (HDFs). In order to study the mechanism(s) by which telomere shortening signals cell senescence, Martens et al. analyzed the telomere length at specific chromosome ends at cumulative population doublings in polyclonal and clonal HDFs by quantitative fluorescence in situ hybridization [38]. The rate of telomere shortening at individual telomeres varied between 50 and 150 bp per population doubling and short telomeres with an estimated 1-2 kb of telomere repeats accumulated prior to senescence. The average telomere length in specific chromosome ends was remarkably similar between clones. However, some exceptions with individual telomeres measuring 0.5-1 kb were observed. In the fibroblast clones, the onset of replicative senescence was significantly correlated with the mean telomere fluorescence but, strikingly, not with chromosomes with the shortest telomere length. The accumulation of short telomeres in late passages of cultured HDFs is compatible with selection of cells on the basis of telomere length and limited recombination between telomeres prior to senescence [38].
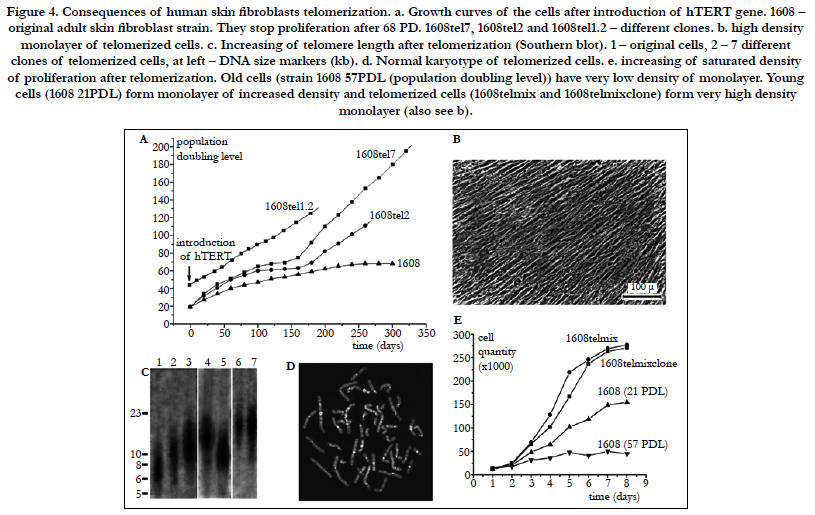
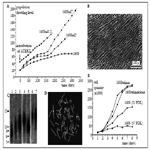
Figure 4. Consequences of human skin fibroblasts telomerization. a. Growth curves of the cells after introduction of hTERT gene. 1608 – original adult skin fibroblast strain. They stop proliferation after 68 PD. 1608tel7, 1608tel2 and 1608tel1.2 – different clones. b. high density monolayer of telomerized cells. c. Increasing of telomere length after telomerization (Southern blot). 1 – original cells, 2 – 7 different clones of telomerized cells, at left – DNA size markers (kb). d. Normal karyotype of telomerized cells. e. increasing of saturated density of proliferation after telomerization. Old cells (strain 1608 57PDL (population doubling level)) have very low density of monolayer. Young cells (1608 21PDL) form monolayer of increased density and telomerized cells (1608telmix and 1608telmixclone) form very high density monolayer (also see b).
It seems that telomeres are responsible for longevity. Recent studies indicate that telomere length, which can be affected by various lifestyle factors, can affect the pace of aging and onset of age-associated diseases (reviewed in ref. [39]). Telomere length shortens with age. Progressive shortening of telomeres leads to senescence, apoptosis, or oncogenic transformation of somatic cells, affecting the health and lifespan of an individual. Shorter telomeres have been associated with increased incidence of diseases and poor survival. The rate of telomere shortening can be either increased or decreased by specific lifestyle factors. Better choice of diet and activities has great potential to reduce the rate of telomere shortening or at least prevent excessive telomere attrition, leading to delayed onset of age-associated diseases and increased lifespan (reviewed in ref. [38]). Our own work highlights the role of telomeres in dermal fibroblasts in skin aging and describes the lifestyle therapeutic nutritional factors including non-hydrolized imidazole containing dipeptide based compounds (non-hydrolized carnosine, carcinine) which may affect telomeres, human health, and skin aging [40-44].
The progressive erosion of the telomere sequence (50–100 bp per mitosis) through successive cycles of replication eventually precludes protection of the ends of the chromosomes, thus preventing end-to-end fusions, which is incompatible with normal cell function. The majority of cells have the capacity for about 60 to 70 postnatal doublings during their lifecycles, and thereafter they reach senescence, remaining viable but incapable of proliferation. This event facilitates end-to-end chromosomal fusions resulting in karyotype disarray with subsequent apoptosis, thus serving as the “biological clock” [34, 45].
Skin aging is affected also by growth factor modifications and hormone activity that declines with age. The best-known decline is that of sex steroids such estrogen, testosterone, dehydroepiandrosterone (DHEA), and its sulfate ester (DHEAS). The effect of this decrease on the skin has been poorly documented, although more data are available for estrogen than for other hormones [46-48]. Other hormones such as melatonin, insulin, cortisol, thyroxine, and growth hormone decline too.
A progressive decrease in androgen production is common in aging men. The physiological causes for this phenomenon seem to be multifactorial [46]. The magnitude of the decline in testosterone with age and the prevalence of older men with low testosterone levels have not been well established. The extent to which an age-dependent decline in androgen levels leads to health problems that might affect or alter the quality of life remains under debate. In men older than middle age, total testosterone levels may be misleading because of an increase in sex hormonebinding globulin levels. The mechanism of the age-associated decrease of the endocrine testicular function is also essentially due to primary testicular failure, but important changes occur at the hypothalamo-pituitary level. The most prominent endocrinological alterations with aging are related to the sex steroids, but others, such as growth hormone, melatonin cortisol, and thyroxine, are also affected. The clinical picture of andropause syndrome is characterized by diminished sexual desire and erectile capacity, decrease in intellectual activity, fatigue, depression, decrease in lean body mass, skin alterations, decrease in body hair, decrease in bone mineral density that results in osteoporosis, and increase in visceral fat and obesity. Clinical response is a better guide to dose requirements, regardless of serum testosterone levels [46].
Aging is associated with a decline in immunity described as immunosenescence. This is paralleled by a decline in the production of several hormones, as typically illustrated by the menopausal loss of ovarian oestrogen production. However, other hormonal changes that occur with aging and that potentially impact on immune function include the release of the pineal gland hormone melatonin and pituitary growth hormone, adrenal production of dehydroepiandrosterone and tissue-specific availability of active vitamin D [48]. At the same time, induced levels of certain signaling molecules such as cytokines and chemokines decline as well, leading to the deterioration of several skin functions [49]. Also, the levels of their receptors decline as well [50]. An elevation of monocyte chemoattractant protein (MCP-1) levels was observed in wounds of aged mice. RNase protection studies, however, revealed that the production of most chemokines, including MIP-2, MIP-1alpha, MIP-1beta, and eotaxin, tended to decline with age [49]. Because optimal wound healing requires both appropriate macrophage infiltration and phagocytic activity, phagocytosis was examined. Compared to young mice, wound macrophages from aged mice exhibited a 37%-43% reduction in phagocytic capacity. Taken together, the data demonstrate age-related shifts in both macrophage and T cell infiltration into wounds, alterations in chemokine content, and a concurrent decline in wound macrophage phagocytic function. These alterations may contribute to the delayed repair response of aging [49]. According to the somatomedin model, growth hormone (GH)-dependent hepatic synthesis is responsible for maintaining circulating insulin-like growth factor (IGF)-I levels. On the other hand, the local autocrine/ paracrine IGF-I expression in peripheral tissue is generally GH-independent and reflects the effects of various and tissuespecific trophic hormones. Circulating IGF-I levels undergo important age-related variations increasing at puberty and decreasing, thereafter, to low levels in the elderly. Low IGF-I levels in the elderly mainly reflect impaired somatotroph secretion but the decline in gonadal sex steroid levels, some protein and micronutrients malnutrition as well as age-dependent variations in IGFbinding proteins may also play a role in the age-related decrease in IGF-I activity [50].
At the same time, some signaling molecules increase with age. One of these is a cytokine called transforming growth factor -beta1, which induces fibroblast senescence. Cellular senescence is a result of molecular alterations in the cellular milieu as well as in DNA and proteins within the cell. All of these changes gradually lead to aberrant cellular response to environmental factors, which can decrease viability and lead to cell death [51].
Replicative senescence of HDFs or melanocytes is caused by the exhaustion of their proliferative potential. Stress-induced premature senescence (SIPS) occurs after many different sublethal stresses including H2O2 (Figure 5), hyperoxia, or tert-butyl hydroperoxide. Cells in replicative senescence share common features with cells in SIPS: morphology, senescence-associated beta-galactosidase activity, cell cycle regulation, gene expression and telomere shortening. Telomere shortening is attributed to the accumulation of DNA single-strand breaks induced by oxidative damage. SIPS could be a mechanism of accumulation of senescent- like cells in vivo. Melanocytes exposed to sublethal doses of UVB undergo SIPS. Melanocytes from dark- and light- skinned populations display differences in their cell cycle regulation. Delayed SIPS occurs in melanocytes from light-skinned populations since a reduced association of p16(Ink-4a) with CDK4 and reduced phosphorylation of the retinoblastoma protein are observed. The role of reactive oxygen species in melanocyte SIPS is unclear. Both replicative senescence and SIPS are dependent on two major pathways. One is triggered by DNA damage, telomere damage and/or shortening and involves the activation of the p53 and p21(waf-1) proteins. The second pathway results in the accumulation of p16(Ink-4a) with the MAP kinase signalling pathway as possible intermediate [52].
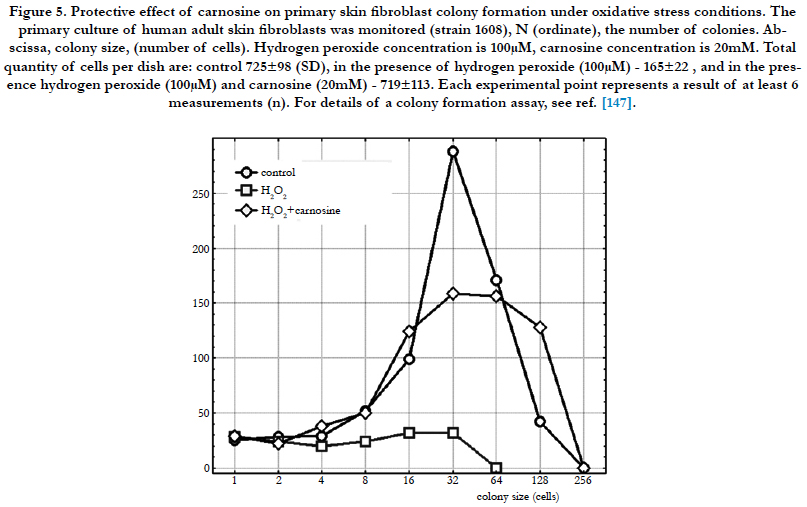

Figure 5. Protective effect of carnosine on primary skin fibroblast colony formation under oxidative stress conditions. The primary culture of human adult skin fibroblasts was monitored (strain 1608), N (ordinate), the number of colonies. Abscissa, colony size, (number of cells). Hydrogen peroxide concentration is 100μM, carnosine concentration is 20mM. Total quantity of cells per dish are: control 725±98 (SD), in the presence of hydrogen peroxide (100μM) - 165±22 , and in the presence hydrogen peroxide (100μM) and carnosine (20mM) - 719±113. Each experimental point represents a result of at least 6 measurements (n). For details of a colony formation assay, see ref. [147].
Normal HDFs exposed to various types of noncytotoxic oxidative stress display a senescent-like phenotype coined SIPS [52]. Such stressful conditions include exposure to hydrogen peroxide [53, 54], tert-butyl hydroperoxide (t-BHP) [55], hyperoxia [56], UV light [57], and radioactivity [58]. Many biomarkers of replicative senescence appear in SIPS: typical cell morphology [55], irreversible growth arrest, lack of response to mitogenic stimuli [54], sharp decrease of the DNA synthesis, and an increase in cells positive for the senescent-associated β-galactosidase activity (SA β-gal) [59].
SIPS is induced 3 days after exposure of HDFs to subcytotoxic oxidative stress with H2O2, with appearance of several biomarkers of replicative senescence [51]. In this work, Frippiat C et al., [51] show that transforming growth factor-beta1 (TGF-beta1) regulates the induction of several of these biomarkers in SIPS: cellular morphology, senescence-associated beta-galactosidase activity, increase in the steady state level of fibronectin, apolipoprotein J, osteonectin, and SM22 mRNA. Indeed, the neutralization of TGF-beta1 or its receptor (TGF-beta RII) using specific antibodies decreases sharply the percentage of cells positive for the senescent-associated beta-galactosidase activity and displaying a senescent morphology. In the presence of each of these antibodies, the steady state level of fibronectin, osteonectin, apolipoprotein J, and SM22 mRNA is no more increased at 72 h after stress [51]. Conditions to induce and parameters to evaluate sublethal oxidative stress of cultured human fibroblasts have been investigated in the attempt to identify markers for a more accurate quantification of cell injury. Sublethal oxidative stress was obtained by treating fibroblasts with 0.5 mM H2O2 in Dulbecco's modified Eagle's medium (DMEM) plus 5% fetal calf serum (FCS) for times not exceeding 60 min. Under these conditions cells remained viable throughout long-term incubation, showing no appreciable release of cytosolic enzymes into the medium. On the contrary, exposures of fibroblasts to 0.5 mM H2O2 for times > 60 min induced a lethal cell injury which was fully expressed 2 days later by massive monolayer wasting and leakage of cytosolic components. Early metabolic effects of sublethal stress consisted of a rapid and significant fall of both ATP and NAD+ pools. Concomitantly, there was a moderate increase (about threefold) in both ADPribosyl transferase activity and free [Ca2+]i, while the specific activity of glyceraldehyde-3-phosphate dehydrogenase was partially decreased upon treatment. Oxidative injury also caused delayed effects consisting of a large depression of both protein and DNA synthesis. However, while the former was partially restored within 10 days of incubation, the latter remained severely impaired, as encountered in a growth-arrested population. Microfilaments of H2O2-treated cells appeared to be morphologically altered due to partial fragmentation of cytoskeleton actin which, however, was still maintained in the polymerized form as F-actin. Moreover, sublethally injured fibroblasts exhibited a reduced adhesiveness to plastic once they were detached and reseeded into new dishes. Relative adhesion efficiencies (number of adherent cells at 16 h as a percentage of seeded cells) were found to correlate inversely with times of exposure to H2O2. This finding allowed the identification of a biological parameter which showed itself to be very sensitive to oxidative stress [53]. Human diploid fibroblast cells lose replicative potential after a certain number of population doublings. Chen and Ames [54] used this experimental system to investigate the role of oxidative damage in cellular aging. Treating cells with H2O2 at < 300 microM did not affect the viability of the majority of cells when judged by morphology, trypan blue exclusion, and protein synthesis. However, the treatment caused a dose-dependent inhibition of DNA synthesis. After a 2-hr treatment with 200 microM H2O2, the cells failed to respond to a stimulus of serum, platelet-derived growth factor, basic fibroblast growth factor, or epidermal growth factor by synthesizing DNA, and the loss of response could not be recovered by 4 days. Subcultivation showed that, as in senescent cells, division of the treated cells was inhibited. The life time cumulative growth curve showed that the loss of replication due to H2O2 treatment was cumulative and irreversible. The H2O2 treatment decreased the number of the population doublings in the rest of the life span by 35.3 +/- 10.3%. Enzymatic assays indicated that, like the cells in their senescent state, the treated cells were less able to activate ornithine decarboxylase and thymidine kinase [54]. Mild oxidative stress as exerted by culture of human WI-38 fibroblasts under 40% oxygen partial pressure blocks proliferation irreversibly after one to three population doublings. Hyperoxically blocked cells are similar to senescent ones in terms of general morphology and lipofuscin accumulation [56]. Moreover, they, like senescent fibroblasts, are blocked preferentially in G1 as evident from DNA content measurements by flow cytometry. Southern blotting of Alu I and Hinf I restricted genomic DNA shows an increase of the rate of telomere shortening from 90 bp per population doubling under normoxia to more than 500 bp per population doubling under hyperoxia. In every case, proliferation is blocked if a telomere cutoff length of about 4 kb is arrived at. The fact that telomere length correlates with the final inhibition of proliferation under conditions of varied oxidative stress, while the population doubling level does not, suggests that telomere shortening provides the signal for cell cycle exit in senescence. In postmitotic cells, no further telomere shortening occurs. However, the sensitivity of terminal restriction fragments to S1 nuclease increases, indicating the accumulation of single-strand breaks in telomeres of nondividing fibroblasts. This effect is found both under normoxic and hyperoxic culture, although it is more pronounced under conditions of higher oxidative stress [56].
Ionizing radiation both in low and high dose ranges exerts differential effects on the cellular processes of differentiation and proliferation in human skin fibroblasts [58]. Single irradiations of fibroblasts in the dose range of 1 to 8 Gy induced terminal differentiation into postmitotic fibrocytes at high percentage level. Irradiation of long-term cultures of fibroblasts with repeated doses of 0.2, 0.6 and 1.0 Gy revealed that only in cultures, which were irradiated repeatedly (x 10) with 0.6 and 1.0 Gy a marked reduction of the proliferation capacity was apparent. Inhibition of proliferation by repeated irradiations with cumulative doses up to 10 Gy was not more pronounced as compared to single irradiations [58].
Dumont and Burton [60] tested the long-term effects of sublethal oxidative stresses on replicative senescence. WI-38 HDFs at early cumulative population doublings (CPDs) were exposed to five stresses with 30 microM I-BHP. After at least 2 d of recovery, the cells developed biomarkers of replicative senescence: loss of replicative potential, increase in senescence-associated beta-galactosidase activity, overexpression of p21(Waf-1/SDI-1/Cip1), and inability to hyperphosphorylate pRb. The level of mRNAs overexpressed in senescent WI-38 or IMR-90 HDFs increased after five stresses with 30 microM I-BHP or a single stress under 450 microM H2O2. These corresponding genes include fibronectin, osteonectin, alpha1(I)-procollagen, apolipoprotein J, SM22, SS9, and GTP-alpha binding protein. The common 4977 bp mitochondrial DNA deletion was detected in WI-38 HDFs at late CPDs and at early CPDs after I-BHP stresses. In conclusion, sublethal oxidative stresses lead HDFs to a state close to replicative senescence [59].
Clinical manifestations of aged skin are xerosis, laxity, wrinkles, slackness, and the occurrence of benign neoplasms such as seborrheic keratoses and cherry angiomas. There are histological features that accompany these changes. In the epidermis, there is no alteration in the stratum corneum and epidermal thickness, keratinocyte shape, and their adhesion, but a decreased number of melanocytes and Langerhans cells is evident [31]. The most obvious changes are at the epidermal-dermal junction: flattening of the rete ridges with reduced surface contact of the epidermis and dermis. This results in a reduced exchange of nutrients and metabolites between these two parts. In the dermis several fibroblasts may be seen, as well as a loss of dermal volume [31,60].
In the study to Lavker et al., [60], the fine structural organization of the epidermis, dermal/epidermal junction, and dermis from an unexposed site (upper inner arm) of elderly people was compared with the organization of a similar region of young people.
Despite an overall thinning of the epidermis and focal areas of cytologic atypia, the characteristic morphological markers associated with the keratinization process are not markedly altered in appearance or amount. A well formed stratum corneum consisting of flattened, enucleated horny cells enveloped by a thickened membrane, and intracellular spaces filled with electron-dense material provide structural evidence that barrier ability is not compromised in senile skin. The dermal/epidermal changes in aged skin are marked and have significant physiologic implications. The major change is a relatively flat dermal/epidermal junction resulting from the retraction of the epidermal papillae as well as the microprojections of basal cells into the dermis. This flattening results in a more fragile epidermal/dermal interface and, consequently, the epidermis is less resistant to shearing forces. Retraction of the epidermal downgrowths (preferential sites of the putative epidermal stem cell) may also explain the loss in proliferative capacity associated with the aged epidermis. The threedimensional arrangements of collagen and elastic fibers showed marked alterations with age. Both fibrous components appear more compact because of a decrease in spaces between the fibers. Collagen bundles appear to unravel, and the individual elastic fibers show signs of elastosis. These changes may contribute to the loss of resilience that is one of the salient features of senile skin [60]. A decrease in blood supply due to a reduced number of blood vessels also occurs. There is also a depressed sensory and autonomic innervation of epidermis and dermis. Cutaneous appendages are affected as well. Terminal hair converts to vellus hair. As melanocytes from the bulb are lost, hairs begin to gray. Further reasons for graying are decreased tyrosinase activity, less efficient melanosomal transfer and migration, and melanocyte proliferation [61]. The primary distinguishing feature of follicular melanogenesis, compared to the continuous melanogenesis in the epidermis, is the tight coupling of hair follicle melanogenesis to the hair growth cycle. This cycle appears to involve periods of melanocyte proliferation (during early anagen), maturation (mid to late anagen) and melanocyte death via apoptosis (during early catagen). Thus, each hair cycle is associated with the reconstruction of an intact hair follicle pigmentary unit... at least for the first 10 cycles or so. Thereafter, gray and white hairs appear, suggesting an age-related, genetically regulated exhaustion of the pigmentary potential of each individual hair follicle. Melanocyte aging may be associated with reactive oxygen species-mediated damage to nuclear and mitochondrial DNA with resultant accumulation of mutations with age, in addition to dysregulation of anti-oxidant mechanisms or pro/anti-apoptotic factors within the cells. While the perception of "gray hair" derives in large part from the admixture of pigmented and white hair, it is important to note that individual hair follicles can indeed exhibit pigment dilution or true grayness. This dilution is due to a reduction in tyrosinase activity of hair bulbar melanocytes, sub-optimal melanocyte-cortical keratinocyte interactions, and defective migration of melanocytes from a reservoir in the upper outer root sheath to the pigmentpermitting microenvironment close to the dermal papilla of the hair bulb [61].
Factors that contribute to wrinkling include changes in muscles, the loss of subcutaneous fat tissue, gravitational forces, and the loss of substance of facial bones and cartilage. Expression lines appear as result of repeated tractions caused by facial muscles that lead to formation of deep creases over the forehead and between eyebrows, and in nasolabial folds and periorbital areas. Repeated folding of the skin during sleeping in the same position on the side of the face contributes to appearance of “sleeping lines.” Histologically, thick connective tissue strands containing muscle cells are present beneath the wrinkle [62]. Skin microrelief alters progressively with age. Wrinkles do not result from these changes but are superimposed upon them. Wrinkles result from structural changes in the epidermis, dermis and hypodermis. Four types of wrinkles can be recognized. Type 1 wrinkles are atrophic. Type 2 wrinkles are elastotic. Type 3 wrinkles are expressional. Type 4 wrinkles are gravitational. Each type of wrinkle is characterized by distinct microanatomical changes and each type of wrinkle develops in specific skin regions. Each is likely to respond differently to treatment. Skin microrelief and skin folds can be identified on histological examination. By contrast, only minimal dermal changes are found beneath permanent or reducible wrinkles compared with immediately adjacent skin [62]. In the muscles an accumulation of lipofuscin (the “age pigment”), a marker of cellular damage, appears. In a study of 80 perioral muscle specimens, lipid pigment (Lipofuscin) granules were present in 68.5% of the cases [63]. The pigmented granules, which were stored at the nuclear poles, were PAS positive, stained black or brown with the Masson-Fontana procedure, black with Sudan black and strong purple-pink with Ziehl-Neelsen staining; yellow autofluorescence was emitted in ultraviolet light. The morphology, histochemical reactions and autofluorescence of the pigmented granules were characteristics of the endogenous lipid pigment--lipofuscin. Statistical analyses indicated a direct correlation between increase in quantity and distribution of the pigment and increase in age (p less than 0.0001), in both males and females [63]. The deterioration of neuromuscular control contributes to wrinkle formation [63].
The constant gravitational force also acts on the facial skin, resulting in an altered distribution of fat and sagging. Skin becomes lax and soft tissue support is diminished. Gravitational effects with advanced years play an important role and contribute to advanced sagging. This factor is particularly prominent in the upper and lower eyelids, on the cheeks, and in the neck region. Fat depletion and accumulation at unusual sites contributes to the altered appearance of the face [64]. It affects the forehead, periorbital, and buccal areas, the inner line of nasolabial folds, and the temporal and perioral regions. At the same time it accumulates submentally, around the jaws, at outer lines of nasolabial folds and at lateral malar areas. In contrast to the young, in whom fat tissue is diffusely distributed, in aged skin fat tends to accumulate in pockets, which droop and sag due to the force of gravity [64,65].
A careful study of the aging face reveals it to be more than just surface textural wrinkling or loose skin. Changes in three-dimensional topography are responsible for the distinctive phenotypic presentation of the face throughout life. These geometric alterations are secondary to apportioning in the fat compartments and result in the fat dysmorphism characteristic of senescence. Redistributing this fat can rebalance the facial fat compartments and mimic the facial structure present in youth [64]. Fat augmentation of the aging face has been a popular cosmetic procedure for decades. Many consider fat the ideal filler because it is natural, nonallergenic, and readily available. However, longevity and symmetry of the fat graft have been unpredictable. In addition, when large volumes are injected for panfacial correction, prolonged edema for months may result. In the study to Butterwick and Lack [65] one hundred patients underwent the F.A.M.I. procedure in which fat was injected within the muscles of facial expression. The fat was harvested in an atraumatic and sterile manner. Centrifuged fat was then injected with specific blunt-tipped cannulae for different muscle groups. The majority of patients underwent a partial- versus full-face procedure. Patients were injected with volumes ranging from 3 to 63 cc of centrifuged fat in a single session. There were no complications, and downtime was 5 to 7 days. Patient satisfaction was high. Patients were followed postoperatively for 3 to 6 months [65].
The mass of facial bones and skeletal bones reduces with age. Resorption affects the mandible, maxilla, and frontal bones. This loss of bone enhances facial sagging and wrinkling with obliteration of the demarcation between the jaw and neck that is so distinct in young persons [66]. It also seems that estrogen and progesterone contribute to elastic fiber maintenance [67]. To confirm this, skin signs and symptoms were examined in 46 menopausal women prior to estrogen replacement therapy. Several symptoms such as pruritus, bruising, dryness and thinning were seen more frequently in sun exposed skin emphasizing the contribution of photoaging [67]. At the end of a 6-mth treatment period, no significant difference was observed in the prevalence or severity of the cutaneous signs and symptoms when patients receiving transdermal 17 beta-estradiol (Estraderm) were compared with controls (the only exception was cutaneous flushing). Elastic fibers from sun protected (buttock) skin of menopausal women were studied by light and electron microscopy. In 3 women (ages 30-37) with a history of premature menopause, the elastic fibers had several degenerative changes including coalescence of cystic spaces into lacunae, peripheral fragmentation, granular degeneration and splitting of the fibers into strands. Similar age-related ultrastructural changes are normally found in individuals that are at least 20 yrs older than these patients. These findings are suggestive of a relationship between premature aging of the dermal elastic fibers and estrogen deprivation [67].
Biomarkers and Factors of Extrinsic Aging
A number of extrinsic, or external, factors often act together with the normal aging process to prematurely age our skin. Most premature aging is caused by sun exposure. Other external factors that prematurely age our skin are repetitive facial expressions, gravity, sleeping positions, and smoking. Extrinsic aging often develops due to several factors: ionizing radiation, severe physical and psychological stress, alcohol intake, poor nutrition, overeating, environmental pollution, and exposure to UV radiation. Among all these environmental factors, UV radiation contributes up to 80%. It is the most important factor in skin aging, especially in premature aging. Both UVB (290–320 nm), and UVA (320–400 nm) are responsible, and the skin alterations caused by UV radiation depend upon the phenotype of photoexposed skin [30,68].
Without protection from the sun rays, just a few minutes of exposure each day over the years can cause noticeable changes to the skin. Freckles, age spots, spider veins on the face, rough and leathery skin, fine wrinkles that disappear when stretched, loose skin, a blotchy complexion, actinic keratoses (thick wart-like, rough, reddish patches of skin), and skin cancer can all be traced to sun exposure.
UVB induces alterations mainly at the epidermal level, where the bulk of UVB is absorbed. It damages the DNA in keratinocytes and melanocytes, and induces production of the soluble epidermal factor (ESF) and proteolytic enzymes, which can be found in the dermis after UV exposure. UVB is responsible for appearance of thymidine dimers, which are also called “UV fingerprints”. That is, after UVB exposure, a strong covalent bond between two thymidines occurs. With aging, this bond cannot be dissolved quickly, and accumulation of mutations occurs. Affected cells appear as sunburn cells 8 to 12 hours after exposure. Reduced production of DNA can be observed during the next 12 hours. Actinic keratoses, lentigines, carcinomas, and melanomas represent delayed effects [3].
“Photoaging” is the term dermatologists use to describe this type of aging caused by exposure to the sun rays. The amount of photoaging that develops depends on: 1) a person’s skin color and 2) their history of long-term or intense sun exposure. People with fair skin who have a history of sun exposure develop more signs of photoaging than those with dark skin. In the darkest skin, the signs of photoaging are usually limited to fine wrinkles and a mottled complexion. Photoaging occurs over a period of years. With repeated exposure to the sun, the skin loses the ability to repair itself, and the damage accumulates. Scientific studies have shown that repeated ultraviolet (UV) exposure breaks down collagen and impairs the synthesis of new collagen. The sun also attacks skin elastin. Sun weakened skin ceases to spring back much earlier than skin protected from UV rays. Skin also becomes loose, wrinkled, and leathery much earlier with unprotected exposure to sunlight. Photoaged skin is characterized by alterations in dermal connective tissue. The skin is increasingly exposed to ambient UV irradiation thus particularly increasing its risk for photooxidative damage with long-term detrimental effects like photoaging, which is characterized by wrinkles, loss of skin tone, and resilience. The amount and structure of this tissue seems to be responsible for wrinkle formation. The dermal extracellular matrix consists of type I and III collagens, elastin, proteoglycans, and fibronectin, and collagen fibrils strengthen the skin. In photoaged skin, collagen fibrils are disorganized and elastin-containing material accumulates [68]. Levels of precursors as well as cross-links between type I and III collagens are reduced, whereas elastin is increased [69,70]. Braverman and Fonferko [69] studied by light and electron microscopy the elastic fibers in he sun exposed and sun protected skin of normal and psoriatic individuals of different ages in order to separate the changes of actinic damage from those of chronological aging. The sun exposed skin showed 2 types of elastic fiber abnormalities-one related to actinic damage and the other to chronological aging. The sun protected buttock skin showed only the latter. From ages 30 to 70, a minority of the elastic fibers exhibited abnormalities that appeared to represent a process of fiber disintegration. After age 70, the majority of elastic fibers showed these abnormalities. These abnormalities were present without accompanying inflammatory cells. Also, there was morphological evidence of continuing synthesis of elastic fibers during the lifetime of these subjects, except that from ages 50-93, the fibers appeared to be loosely, rather than compactly, assembled [69]. Talwar et al., [70] quantitatively assessed the relation between type I and type III procollagen precursor levels and the severity of clinical photodamage in human skin. Levels of procollagen, pN collagen (collagen without the carbroxypropeptide), and/or pC collagen (collagen without the aminopropeptide) were determined by radioimmunoassay, Western blot, and immunohistology in punch biopsy specimens from mildly and severely photodamaged forearm skin and from sunprotected underarm and buttock skin of the same subjects. Collagen precursor levels in forearm and underarm skin were expressed relative to buttock levels for comparison. In the mildly photodamaged group, collagen precursors in the forearm did not differ from those in the underarm by any measurement, except for type I collagen precursors measured by radioimmunoassay, which were reduced 16%. In severely photodamaged forearm skin, both type I and type III collagen precursor levels, measured by radioimmunoassay, were significantly reduced (approximately 40%).
Western analysis revealed similar significant reductions in type I and type III collagen precursor levels in severely photodamaged forearm skin compared with the sun-protected underarm. Immunohistology localized both type I and III pN collagens predominantly to the extracellular papillary dermis. Relative staining intensities of type I and type III pN collagen were also significantly reduced in severely photodamaged forearm skin. Multiple linear regression modeling of all data demonstrated that reductions in collagen precursor levels were significantly correlated (p < 0.03) with the severity of photodamage, but not with chronologic age. These data demonstrate, by three independent methods, coordinate reductions of both type I and type III collagen precursors in photodamaged human skin, and the degree of reduction correlated with the degree of photodamage. It is likely that such changes in collagen precursors lead to reduced levels and/or altered organization of fibrillar collagen, and thus may contribute to the wrinkled appearance of photodamaged human skin [70]. Type I and type III procollagen are reduced in photodamaged human skin. This reduction could result from increased degradation by metalloproteinases and/or from reduced procollagen synthesis. In the separate study, Varani et al., [71] investigated type I procollagen production in photodamaged and sun-protected human skin. Skin samples from severely sun-damaged forearm skin and matched sun-protected hip skin from the same individuals were assessed for type I procollagen gene expression by in situ hybridization and for type I procollagen protein by immunostaining. Both mRNA and protein were reduced (approximately 65 and 57%, respectively) in photodamaged forearm skin compared to sun-protected hip skin. We next investigated whether reduced type I procollagen production was because of inherently reduced capacity of skin fibroblasts in severely photodamaged forearm skin to synthesize procollagen, or whether contextual influences within photodamaged skin act to down-regulate type I procollagen synthesis. For these studies, fibroblasts from photodamaged skin and matched sun-protected skin were established in culture. Equivalent numbers of fibroblasts were isolated from the two skin sites. Fibroblasts from the two sites had similar growth capacities and produced virtually identical amounts of type I procollagen protein. These findings indicate that the lack of type I procollagen synthesis in sun-damaged skin is not because of irreversible damage to fibroblast collagen-synthetic capacity. It follows, therefore, that factors within the severely photodamaged skin may act in some manner to inhibit procollagen production by cells that are inherently capable of doing so. Interactions between fibroblasts and the collagenous extracellular matrix regulate type I procollagen synthesis. In sun-protected skin, collagen fibrils exist as a highly organized matrix. Fibroblasts are found within the matrix, in close apposition with collagen fibers. In photodamaged skin, collagen fibrils are shortened, thinned, and disorganized. The level of partially degraded collagen is approximately 3.6-fold greater in photodamaged skin than in sun-protected skin, and some fibroblasts are surrounded by debris. To model this situation, skin fibroblasts were cultured in vitro on intact collagen or on collagen that had been partially degraded by exposure to collagenolytic enzymes. Collagen that had been partially degraded by exposure to collagenolytic enzymes from either bacteria or human skin underwent contraction in the presence of dermal fibroblasts, whereas intact collagen did not. Fibroblasts cultured on collagen that had been exposed to either source of collagenolytic enzyme demonstrated reduced proliferative capacity (22 and 17% reduction on collagen degraded by bacterial collagenase or human skin collagenase, respectively) and synthesized less type I procollagen (36 and 88% reduction, respectively, on a per cell basis).
Taken together, these findings indicate that 1) fibroblasts from photoaged and sun-protected skin are similar in their capacities for growth and type I procollagen production; and 2) the accumulation of partially degraded collagen observed in photodamaged skin may inhibit, by an as yet unidentified mechanism, type I procollagen synthesis [71].
Photoaged skin displays prominent alterations in the cellular component and the extracellular matrix of the connective tissue with an accumulation of disorganized elastin and its microfibrillar component fibrillin and other constituents of elastin-associated microfibrils in the deep dermis, and a severe loss of interstitial collagens, the major structural proteins of the dermal connective tissue. The unifying pathogenic agents for these changes are UV-generated ROS that deplete and damage non-enzymatic and enzymatic antioxidant defense systems of the skin. As well as causing permanent genetic changes, ROS activate cytoplasmic signal transduction pathways in resident fibroblasts that are related to growth, differentiation, senescence, and connective tissue degradation [68]. It is accepted that UVA radiation plays an important role in the pathogenesis of photoaging [72]. UVA penetrates more deeply into the dermis and damages both the epidermis and dermis. The amount of UVA in ambient light exceeds the UVB by 10 to 100 times, but UVB has biological effects 1,000 times stronger than UVA [3]. There is increasing evidence for the generation of ROS in skin upon ultraviolet exposure, but little is known about their pathophysiologic relevance in human skin in vivo. Sander et al., [73] proposed that chronic and acute photodamage is mediated by depleted antioxidant enzyme expression and increased oxidative protein modifications. Biopsies from patients with histologically confirmed solar elastosis, from non-ultraviolet-exposed sites of age-matched controls, and from young subjects were analyzed. To evaluate the influence of acute ultraviolet exposures, buttock skin of 12 healthy subjects was irradiated repetitively on 10 d with a solar simulator and compared intraindividually to non-ultraviolet treated contralateral sites. The antioxidant enzymes catalase, copper-zinc superoxide dismutase, and manganese superoxide dismutase were investigated by immunohistochemistry. Protein carbonyls were analyzed by immunohistochemical and immunoblotting techniques in human skin and in cell models. Whereas overall expression of antioxidant enzymes was very high in the epidermis, low baseline levels were found in the dermis. In photoaged skin, a significant depletion of antioxidant enzyme expression was observed within the stratum corneum and in the epidermis. Importantly, an accumulation of oxidatively modified proteins was found specifically within the upper dermis of photoaged skin. Upon acute ultraviolet exposure of healthy subjects, depleted catalase expression and increased protein oxidation were detected. Exposures of keratinocytes and fibroblasts to ultraviolet B, ultraviolet A, and H2O2 led to dosedependent protein oxidation and thus confirmed in vivo results [73]. A decrease in the overall ROS load by efficient sunscreens or other protective agents may represent promising strategies to prevent or at least minimize ROS induced photoaging [68].
UV induces the matrix metalloproteinases collagenase, 92-kD gelatinase, and stromelysin, which degrade skin connective tissue and may contribute to premature skin aging (photoaging) [74]. UV radiation increases the production of collagen-degrading enzymes, matrix metalloproteinases (MMPs), and the xeroderma pigmentosum factor (XPF), which can also be found in the epidermis. XPF induces epidermal-dermal invagination, representing the beginning of wrinkle formation. At the base of wrinkles, less type IV and VII collagen is found. This instability deepens the wrinkles. Each MMP degrades a different dermal matrix protein; for example, MMP-1 cleaves collagen types I, II, and III, and MMP-9 (gelatinase) degrades type IV and V and gelatin. Under normal conditions, MMPs are part of a coordinated network and are regulated by their endogenous inhibitors (TIMPs). The imbalance between activation and inhibition can lead to proteolysis [74].
Chronic sun exposure can result in numerous changes in human skin, particularly the skin of fair-skinned individuals. These changes include wrinkling, elastosis, actinic keratoses, irregular pigmentation, telangiectasia, and skin cancer. Most of these alterations had formerly been considered to be caused by UVB wavelengths, which are the wavelengths probably most responsible. However, a number of recent studies have demonstrated that UVA can also cause burning, elastosis, and skin cancer [75]. The activation of MMPs can be triggered by UVA and UVB, but molecular mechanisms differ depending upon the type of radiation. UVA radiation can generate ROS that affect lipid peroxidation and generate DNA strand breaks [75]. On the other hand, within minutes after exposure UVB radiation causes MMP activity and DNA damage. These effects can be observed after exposing human skin to one-tenth of the minimal erythema dose.
Another environmental factor contributing to premature aging is smoking. Tobacco smoking is a serious and preventable health hazard that can cause or exacerbate a number of diseases and shorten life expectancy, but the role of smoking as an etiologic factor in the development of skin disease is largely unknown. Tobacco smoke contains a complex mixture of gaseous and particulate compounds, several of which may have the potential to exert physiologic and pharmacologic impacts (Figure 6). Nicotine has for decades been regarded as a primary factor that engenders smoking-related disorders, but recent evidence clearly demonstrates that its temporary vasoactive effect on the skin and subcutaneous perfusion cannot satisfactorily explain the pathophysiologic mechanisms that impair wound healing and contribute to smoking-related disorders [76]. Although epidemiological evidence is sparse, findings suggest that tobacco smoking is a contributing factor in systemic lupus erythematosus, psoriasis, palmoplantar pustulosis, cutaneous squamous cell carcinoma, hidradenitis suppurativa, and genital warts [77]. In contrast, smoking may confer some protective effects and mitigate other skin diseases, notably pemphigus vulgaris, pyoderma gangrenosum, aphthous ulcers, and Behçet's disease. Various degenerative dermatologic conditions are also impacted by smoking, such as skin wrinkling and dysregulated wound healing, which can result in post-surgical complications and delayed or even arrested healing of chronic wounds. Most likely, alteration of inflammatory cell function and extracellular matrix turnover caused by smoking-induced oxidative stress are involved in the pathophysiologic mechanisms [77].
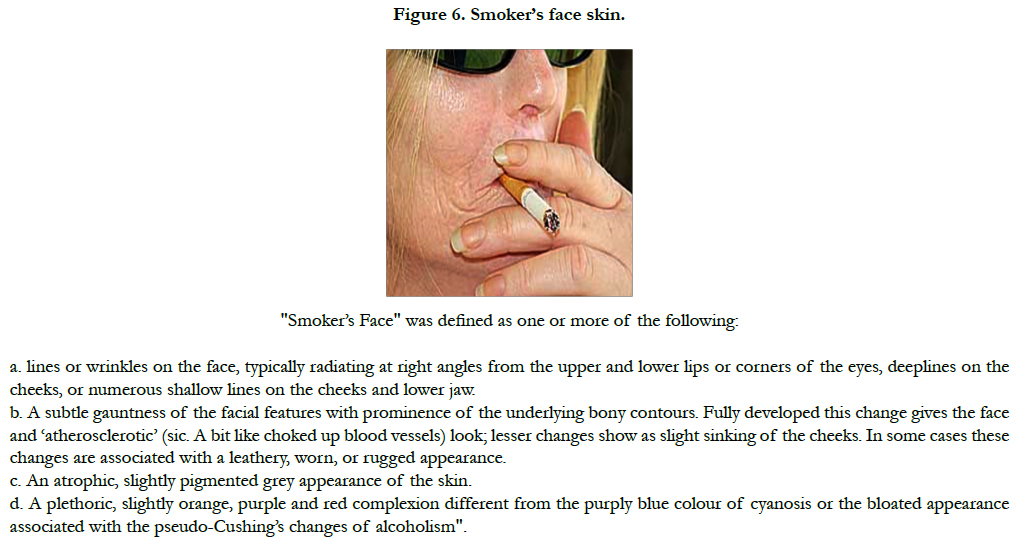
Figure 6. Smoker’s face skin.
“Smoker’s face” or “cigarette skin” are characteristic, implying increased facial wrinkling and an ashen and gray skin appearance [78]. Cigarette smoking is strongly linked to serious internal diseases such as cancer, cardiovascular disease, and lung disease. However, the external manifestations and consequences of smoking are relatively unknown. Although generally less ominous, the cutaneous manifestations of smoking may be associated with significant morbidity [77]. Smoking produces systemic immunomodulatory effects through the release of ROS from tobacco smoke, which is believed to cause a cascade of detrimental effects on normal inflammatory cell function by attenuating phagocytosis and bactericidal mechanisms, as well as by increasing the release of proteolytic enzymes [79, 80]. In addition, collagen synthesis and the deposition of mature collagen in the extracellular matrix are reduced through smoking [81]. Because mature collagen is the main determinator of strength of an operative wound, the results support the view that patients should be advised to stop smoking before an operation [81]. Such disruptive influences on these biologic mechanisms culminate in adverse affects on the cellular reparative pathways of the skin and its appendages, which can be observed in the healing of acute wounds in smokers. This cohort has a significantly higher risk of post-operative dehiscence and infection, and in patients who are heavy smokers, slowed or arrested healing of chronic wounds can also occur [82]. Undoubtedly, degenerative skin disorders are the result of smokinginduced defects in reparative mechanisms and the progression of extracellular degradation of elastin, collagen, and other extracellular matrix molecules. More complex, and in fact unknown, is the role of smoking in the etiology of autoimmune and neoplastic skin diseases. It is increasingly apparent that the immunomodulatory effects and alteration of inflammatory cell function from smoking influences the clinical course of cutaneous diseases. Dermatologic research still needs to elucidate why smoking is an aggravating factor for some diseases, while appearing to mitigate the clinical course of others. A prematurely old appearance is a symptom of long-term smokers. Yellow and irregularly thickened skin is result of elastic tissue breakdown due to smoking [83] or to UV. While strategies for framing messages about the association between smoking and facial aging await further study, this association deserves to be considered in all tobacco control and counteradvertising campaigns. Although the tobacco industry promotes images of glamour, 2 decades of epidemiologic research have concluded the opposite: smokers have enhanced facial aging and skin wrinkling compared with nonsmokers [83]. Although there are methodological concerns with each of these studies, the data are consistent with the conclusion that smoking causes skin wrinkling that could make smokers appear unattractive and prematurely old [84]. Cigarette smoking has been shown to decrease capillary and arteriolar blood flow in the skin, perhaps damaging connective tissue components that are important to maintaining the integrity of the skin. Americans are highly motivated to avoid or eliminate facial wrinkles. The association of smoking and facial wrinkling may be important evidence to convince young persons not to begin smoking and older smokers to quit [84]. It seems that cigarette smoking induces the activation of MMPs in the same mode as in persons with significant sun exposure [85]. Lahmann et al., [85] have compared the concentrations of mRNA for matrix metalloproteinase 1 (MMP-1) in the buttock skin of smokers and non-smokers with quantitative real-time polymerase chain reactions. MMP-1 degrades collagen, which accounts for at least 70% of the dry weight of dermis. We report significantly more MMP-1 mRNA in the skin of smokers than non-smokers whereas no difference was seen for the tissue inhibitor of metalloproteinases 1 (TIMP-1) or the housekeeping gene GAPDH (glyceraldehyde- 3-phosphate dehydrogenase). The authors suggest that smokinginduced MMP-1 might be important in the skin ageing effects of tobacco smoking [85]. Smoking also reduces facial stratum corneum moisture as well as vitamin A levels, which is important in reducing the extent of collagen damage [30].
The photochemical activity of smog is due to the reduction of air pollutants such as nitrogen oxides and volatile organic compounds created from fossil fuel combustion in the presence of sunlight. Emission from factories and motor vehicle exhaust are primary sources of these compounds. The major targets of ozone in the skin are the superficial epidermal layers; this results in the depletion of antioxidants such as alpha-tocopherol (vitamin E) and ascorbic acid (vitamin C) in the superficial epidermal layers [86].
As stochastic damage is explained, the damage is initiated by random cosmic radiation and triggered by free radicals during cell metabolism, which damages cell lipid compounds, especially membrane structures [3, 87].
Therapeutic Solutions to Improve Aging Skin. Innovative Oral Therapeutic Dosage Forms of Non-Hydrolized Carnosine and Carcinine are Acting for Skin Beautification and Rejuvenation Through the Protective Telomere -Mediated Treatment Effects on Dermal Fibroblasts
Medical and procedural interventions are available to improve aging skin (reviewed in ref. [12]). Medical interventions typically refer to topical agents that will prevent sun damage or stimulate the production of new collagen. These treatments are long-term solutions, and the results, although measurable, can be subtle and occur over fairly long periods of time. Medical treatments focus mainly on the issue of abnormal collagen; such treatments include the use of topical retinoids, select antioxidants, and, more recently, drugs that are used primarily to resolve precancerous skin lesions known as actinic keratoses. Medical treatments are usually in the form of prescription medications, but some are available as over-the-counter preparations.
Procedural treatments, on the other hand, tend to lead to more dramatic changes, and the effects are noted sooner than with medical therapy alone. These procedures are often employed in conjunction with medical therapy to combat the effects of aging skin. Procedures such as laser surgery can address dyspigmentation, redness, and textural changes. Soft tissue fillers address the volume loss associated with aging. Botulinum toxin injections are used to treat dynamic wrinkles associated with repeated muscle movement and that ultimately lead to “fixed” wrinkles at rest. These procedures are administered by trained professionals and can be associated with significant side effects [12].
A growing body of research is showing how nutricosmetics can contribute to healthy skin. While topical creams and cosmetic products can affect skin condition from the outside, nutritional supplements taken orally can have an impact from within the skin: this is called “Beauty from Within”.
Carnosine (β-alanyl-L-histidine) and related compounds are natural constituents of excitable tissues possessing diverse biological activities [88, 89]. The level of carnosine in tissues is controlled by a number of enzymes transforming carnosine into other carnosine related compounds, such as carcinine, N-acetylcarnosine, anserine or ophidine (by decarboxylation, acetylation or methylation, respectively) or its cleavage into the amino acids, histidine and β-alanine. Hydrolysis is mainly due to tissue carnosinase (EC 3.4.13.3) which is widely distributed among different subjects [44, 90, 91] or serum carnosinase (EC 3.4.13.20), present in brain and blood plasma of primates and humans [44, 92, 93]. Carnosine has been proven to scavenge ROS as well as alpha-beta unsaturated aldehydes formed from peroxidation of cell membrane fatty acids during oxidative stress [94, 95]. It can oppose glycation [96, 97] and it can chelate divalent metal ions [98]. The important studies have produced clinical and experimental evidence of beneficial effects of N-acetylcarnosine in treating cataracts of the eyes, these and other ophthamological benefits have been proven [99- 109]. Carcinine (β-alanyl histamine) is an imidazole dipeptide first discovered in the crustacean Carcinus maenas [Reviewed in refs. [44, 89, 110] , and has subsequently been found in the hearts of several mammalian species [111, 112]. It has been demonstrated that carcinine is metabolically related to histamine, histidine, and carnosine (β-alanyl histidine), and could be synthesized from histamine and β-alanine [113]. In addition, previous studies have shown that carcinine contains an imidazole group with flexible ethylene side chain known to be important for histamine H3 receptor-ligand interactions [114-117]. From these findings, it seems that a certain relationship exists between brain histamine and carcinine, and that carcinine might be a new histamine H3 receptor antagonist. The results of the recent study provide direct evidence that carcinine, as a novel histamine H3 receptor antagonist, plays an important role in histaminergic neurons activation and might be useful in the treatment of certain diseases, such as epilepsy, and locomotor or cognitive deficit [118]. In two of the reports, carcinine was shown to act as a natural antioxidant [119, 120] and to play a role in regulating stress and shock with a 1000-fold less potent hypotensive effect than histamine [112, 121], suggesting that carcinine might have therapeutic use (reviewed in Ref. [122, 123]). Overall, these low molecular mass antioxidant peptidomimetics add significantly to the host defense provided by the enzymes superoxide dismutase, catalase and glutathione peroxidases and act as pharmacological chaperones in biological systems [44, 89, 119, 120, 123].
Innovative Vision Products, Inc. (IVP) metabolic scientists have validated an oral drug delivery platform of non-hydrolized carnosine and are in the process of validating a drug delivery platform for carcinine which could be used to redesign existing injected peptides to enable oral uptake. This technology is based on an understanding of the dipeptide structure stabilization from the enzymatic hydrolysis with carnosinases during the oral delivery of a peptide drug which was found to be inherently orally available (can be swallowed rather than injected or topically applied to the skin with a cream) [40, 41]. The vast majority of peptides (fragments of proteins, or very small proteins) are broken apart by digestive enzymes or acid in the stomach and intestines before they have a chance to be absorbed into the body tissues, so most peptide drugs cannot be taken by the patient preferred oral route and are usually injected. The injected route of administration usually limits the commercial value of peptide drugs, even though they can be very effective drugs with good safety and tolerability profiles, because their design is often based on natural molecules. The ability to convert any of these peptides to oral drugs could have significant commercial value. Delivery of peptide drugs or peptide cosmetic ingredients through the skin is an attractive alternative to needles, but has proved elusive thus far. The rewards for effective transdermal drug delivery of peptides are large. Drug delivery using skin patches has grown into a multibillion dollar industry, with multiple commercial and clinical successes for a variety of small drugs. Patients like patches because of their convenience - there is no need to remember to take frequent pills and no pain from hypodermic injections. Doctors like patches because of their efficacy - transdermal delivery avoids the complications of poor absorption and enzymatic degradation associated with oral delivery and eliminates the peaks and valleys of drug concentration in the blood associated with bolus injections. And pharmaceutical companies like patches because of their profitabilitypatches are not only preferred by their customers but can often be used, in effect, to extend the patent life of a drug through new and improved delivery. These advantages have motivated the research community to overcome the primary challenge of transdermal delivery: however, the skin's outer layer of stratum corneum is an extremely tough barrier for peptide compounds, that generally only permits entry of small, lipophilic drugs and uniformly excludes large, hydrophilic proteins. Various chemical enhancers, such as surfactants, have been used to increase skin permeability to small molecules but, with few exceptions, have been ineffective in delivering proteins. Physical approaches, which breach the skin's barrier more aggressively, have recently met with some success for proteins and other compounds. Electric fields, ultrasound and jet injectors have received US Food and Drug Administration approval for transdermal applications, and microneedles and thermal ablation are being studied in clinical trials. Although these methods show promise, they may be limited by the need for devices that could be large, costly and cumbersome [123, 124].
A range of candidate compounds in the oral formulation of nonhydrolized carnosine have been screened [40-43, 125, 126] and fine-tuned to identify the most promising candidates for potential development as an orally dosable drug or therapeutic nutritional product for the treatment of skin aging supported by morphometry (computerized image analysis). Skin aging is usually accompanied by the increase with age of the surface density of elastin fibers and by a corresponding decrease of elasticity [126]. The clinical data with the oral formulations of non-hydrolized carnosine for the skin beautification provide proof-of-concept therapeutic platform that tackles these disturbances of the skin and application of this technology to other interesting biomedical applications is now underway [40-43, 125, 126]. To assess the efficacy and acceptability of the oral products including imidazolecontaining dipeptide based compounds (non-hydrolized carnosine or carcinine), a double-blind randomized clinical study versus placebo was carried out [126]. A 4-month randomized doubleblind controlled study was undertaken including 42 subjects where the oral supplement of non-hydrolized carnosine (Can-C Plus® formulation) (Figure 7) was tested against placebo for 3 months followed by a 1-month supplement-free period for both groups to assess lasting effects. Assessment of the age-related skin parameters and oral treatment efficacy measurements included objective skin surface evaluation with Visioscan® VC 98 and visual assessment of skin appearance parameters. The results together confirm that a direct 1-electron transfer between a Schiff base methylglyoxal dialkylimine (or its protonated form) and methylglyoxal is responsible for the generation of the cross-linked radical cation and the radical counteranion of methylglyoxal. Under aerobic conditions, molecular oxygen can then accept an electron from the methylglyoxal anion to generate the superoxide radical anion causing the propagation of oxidative stress chain reactions in the presence of transition metal ions. Carnosine stabilized from the enzymatic hydrolysis, arcinine and leucyl histydylhidrazide in patented formulations thereof demonstrate the Schiff bases transglycating activities concomitant with glycation site specific antioxidant activities and protection of proprietary antioxidant enzymes in the skin during aging and diabetes lesions. During oral supplementation of stabilized from enzymatic hydrolysis carnosine (Can-C Plus® formulation), skin investigated parameters showed a continuous and significant improvement in the active group during the 3 months of supplementation as compared to placebo. Visual investigation showed improvement of the overall skin appearance and reduction of fine lines. No treatment- related side effects were reported [126]. The finding that already-formed Advanced glycation Maillard reaction end products (AGE) crosslinks can be pharmacologically severed and attendant pathology thereby reversed by non-hydrolized carnosine or carcinine in patented oral formulations thereof has broad implications for the skin beautification and therapeutics of the complications of diabetes and skin diseases associated with aging. A striking anti-senescence effect of carnosine was demonstrated by McFarland and Holliday [127-129]. They showed that human diploid fibroblasts grown in 20 mM carnosine had an extended lifespan, both in population doublings and chronological time. The published study [127] has detalized that dipeptide L-carnosine has beneficial effects on cultured human fibroblasts. Physiological concentrations in standard media prolong their in vitro lifespan and strongly reduce the normal features of senescence. Late passage cells in normal medium are rejuvenated when transferred to medium containing carnosine, and become senescent when carnosine is removed. In the absence of pyruvate, carnosine is cytotoxic to neoplastic and transformed human and rodent cells. Mc- Farland and Holliday [128] have examined the effects of the naturally occurring dipeptide carnosine (beta-alanyl-L-histidine) on the growth, morphology, and lifespan of cultured HDFs. With humanforeskin cells, HFF-1, and fetal lung cells, MRC-5, McFarland and Holliday have shown that carnosine at high concentrations (20-50 mM) in standard medium retards senescence and rejuvenates senescent cultures. These late-passage cultures preserve a nonsenescent morphology in the presence of carnosine, in comparison to the senescent morphology first described by Hayflick and Moorhead (reviewed in ref. [128]). Transfer of these latepassage cells in medium containing carnosine to unsupplemented normal medium results in the appearance of the senescent phenotype. The serial subculture of cells in the presence of carnosine does not prevent the Hayflick limit to growth, although the lifespan in population doublings as well as chronological age is often increased. This effect is obscured by the normal variability of human fibroblast lifespans, which McFarland and Holliday have confirmed. Transfer of cells approaching senescence in normal medium to medium supplemented with carnosine rejuvenates the cells but the extension in lifespan is variable. Neither D-carnosine, (beta-alanyl-D-histidine), homocarnosine, anserine, nor beta-alanine had the same effects as carnosine on human fibroblasts. Carnosine is an antioxidant, but it is more likely that it preserves cellular integrity by its effects on protein metabolism. The muscle and brain-associated dipeptide, carnosine, is an intracellular buffer which can delay senescence in cultured human fibroblasts and delay aging in senescence-accelerated mice. The effect of advanced glycation end products (AGE-s) was studied on the proliferation and cell death of human skin fibroblasts in culture. Several AGEproducts were prepared from proteins, a peptide and amino acids, using Glucose or Fructose, with or without Fe2+. The AGE preparations increased cell death at the 7th day, after only 72 hours of incubation. Some of these glycation products modified also proliferation. This effect of AGE-s was even maintained without these products in fresh medium for a second period of incubation up to 10 days from the start of the experiment. In order to explore the role of AGE-receptors, especially of AGE-receptor and of growth factor receptors (fibroblast and epidermal growth factors receptors), antibodies to these receptors were added to cell cultures and their effect on both cell death and proliferation were determined as for the AGE-s. These anti-receptor antibodies imitated to some extent the results obtained with AGE-s, producing increase of cell death and proliferation, followed above a certain concentration of antibodies by a decrease and a new increase or plateau. This might correspond to the internalization of the receptors followed by a re-expression on the cell membrane. The role of receptor-mediated ROS- production was also explored using scavengers: N-acetyl-cysteine (NAC), L-Carnosine, superoxide dismutase (SOD) and Catalase. Several of these scavengers decreased cell death, suggesting that ROS-production is partially involved in the observed phenomena [130]. Carnosine's ability to react with methylglyoxal (MG) and possibly other deleterious carbonyl compounds, and scavenge various ROS, may account for its protective ability towards ischemia and ageing [126, 131]. At the cellular level 20 mM carnosine protected cultured human fibroblasts and lymphocytes, Chinese hamster ovary (CHO) cells, and cultured rat brain endothelial cells against the toxic effects of formaldehyde, acetaldehyde and malonic dialdehyde (MDA), and AGEs formed by a lysine/deoxyribose mixture [132]. A mouse model was used to investigate whether tumor growth in vivo can be inhibited by carnosine. Therefore, NIH3T3 fibroblasts, conditionally expressing the human epidermal growth factor receptor 2 (HER2/neu), were implanted into the dorsal skin of nude mice, and tumor growth in treated animals was compared to control mice [133]. In two independent experiments nude mice that received tumor cells received a daily intra peritoneal injection of 500 microl of 1 M carnosine solution. Measurable tumors were detected 12 days after injection. Aggressive tumor growth in control animals, that received a daily intra peritoneal injection of NaCl solution started at day 16 whereas aggressive growth in mice treated with carnosine was delayed, starting around day 19. A significant effect of carnosine on tumor growth was observed up to day 24. Although carnosine was not able to completely prevent tumor growth, a microscopic examination of tumors revealed that those from carnosine treated animals had a significant lower number of mitosis (p < 0.0003) than untreated animals, confirming that carnosine affects proliferation in vivo. As a naturally occurring substance with a high potential to inhibit growth of malignant cells in vivo, carnosine should be considered as a potential anti-cancer drug [133]. Two-dimensional electrophoretic gel profiles were compared between rat 3Y1 fibroblasts cultured in the presence and absence of 30 mM L-carnosine (beta-alanyl-L-histidine) for one week without any replenishment of medium. While a number of cellular proteins changed their expression levels by the addition of carnosine, Ikeda et al., [134] identified one of the most prominently varied proteins as vimentin. Immunoblot analysis with anti-vimentin antibody demonstrated that the vimentin levels increased about 2-fold after one-week culture in the presence of carnosine. Ikeda et al., [134] also confirmed that the increase of vimentin expression was dependent on the concentration of carnosine added to the medium. Moreover, when cultured cells were stained with anti-vimentin antibody and observed by light microscopy, most cells grown in the presence of carnosine were found to have markedly developed vimentin filaments. The increase of vimentin expression was also observed by adding with carnosine related dipeptides, N-acetylcarnosine and anserine [134]. Using three different experimental approaches (microscopic, flow cytometric and ELISA for one of the markers of DNA oxidative damage) the study on rat embryonic fibroblasts [135] has demonstrated that L-carnosine at 30 mM concentration sustains the retention of cell morphology even during a nutritional insult for five weeks. Also, L-carnosine significantly reduces the formation of 8-hydroxy deoxyguanosine (8-OH dG) in the cells after four weeks of continuous culture. Thus it could be inferred that the anti-senescent effect of L-carnosine to fibroblasts is probably linked to its inhibition of formation of intracellular 8-OH dG during oxidative stress [135]. An enhancing effect of L-histidine (L-His) was detected on the induction by hydrogen peroxide (H2O2) of chromosomal aberrations of both the chromosome type and the chromatid type, in human embryonic fibroblasts [136]. The maximum efficiency of induction was about 8-fold higher in the presence of L-His than in the presence of H2O2 alone, at a concentration of L-His of 50 microM. D-His and DL-His showed lower enhancing effects than L-His, with approximately 2-fold and 5-fold enhancement of induction of chromosomal aberrations, respectively. L-Histidinol and L-His-methyl ester, among various derivatives of L-His tested, also enhanced this process. However, the effects of these derivatives were smaller than those of L-His in a range of concentrations equivalent to that of the most effective dose of L-His (50 microM), while they produced greater enhancement than L-His at concentrations higher than 200 microM. The results indicate that at least both the imidazole ring and the amino group are essential components of the chemical structure of L-His required for the enhancing effect. Moreover, in order to cause such an enhancing effect, L-His had to be applied together with H2O2 to cells, because the enhancing effect of L-His was not observed with cells which were washed after pretreatment with L-His. The preliminary study suggested that this enhancing effect depends on the His-peroxide adduct derived from L-His and H2O2. None of the amino acids tested other than His produced any enhancing effect on the induction of chromosomal aberrations by H2O2 [136].
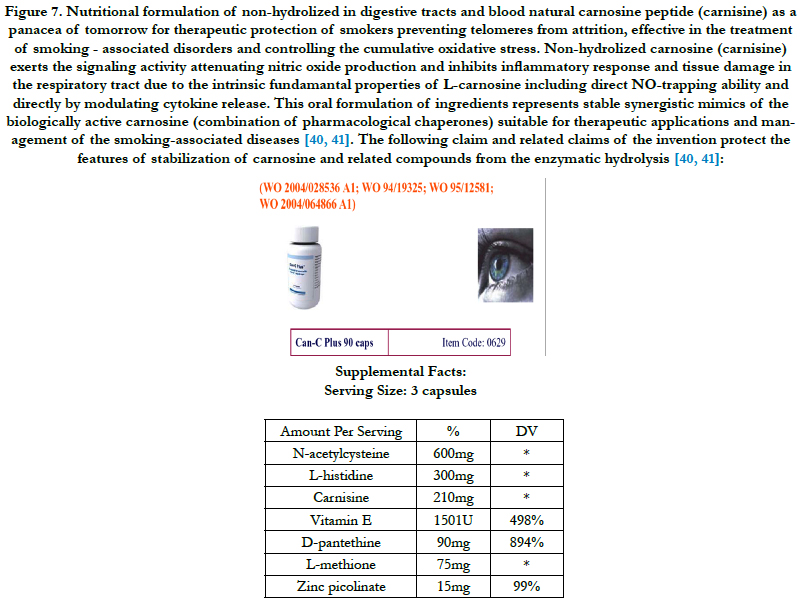

Figure 7. Nutritional formulation of non-hydrolized in digestive tracts and blood natural carnosine peptide (carnisine) as a panacea of tomorrow for therapeutic protection of smokers preventing telomeres from attrition, effective in the treatment of smoking - associated disorders and controlling the cumulative oxidative stress. Non-hydrolized carnosine (carnisine) exerts the signaling activity attenuating nitric oxide production and inhibits inflammatory response and tissue damage in the respiratory tract due to the intrinsic fundamantal properties of L-carnosine including direct NO-trapping ability and directly by modulating cytokine release. This oral formulation of ingredients represents stable synergistic mimics of the biologically active carnosine (combination of pharmacological chaperones) suitable for therapeutic applications and management of the smoking-associated diseases [40, 41]. The following claim and related claims of the invention protect the features of stabilization of carnosine and related compounds from the enzymatic hydrolysis [40, 41]:
Above Figure Description:
% Daily Value * not established Other ingredients: Rice flour, magnesium sterate, vegetable capsule. Note: Keep in cool dark conditions, out of the reach of children and consume before end of expiry date. Not for use by pregnant or lactating women.
Use of a combination of a carnosinase inhibitor with L-carnosine or its related substance which is a substrate of carnosinase, for manufacturing a pharmaceutical or nutritional composition for increasing the carnosine or its related substance in plasma or cells of mammals.
wherein at least one of the carnosinase inhibitors is used selected from the group comprising of:
1. β-alanine
2. β-alanine comprising compound,
3. an N-alkanoyl-β-alanine compound
4. N-alkanoyl-L-carnosine compound
5. bestatin presented by the pharmaceutically acceptable formula of bestatin
6. an α-amino acid derivative acting as inhibitor of carnosinase activity and/or a dipeptidase activity, specific to hydrolize β-alanylcontaining dipeptides (carnosine related compounds),
7. a γ- amino acid derivative, acting as carnosinase inhibitor of a dipeptidase activity, specific to hydrolize β-alanyl-containing dipeptides(carnosine related compounds),v
8. a thiol containing reducing agent essentially comprising unithiol, dithiothreitol, 2 mercaptoethanol, N-acetylcysteine or related compounds acting as a thiol type reducing agent,
9. a thiol antioxidant,
10. a metal chelating agent,
11. a hydrophobic compound with a branched hydrocarbon residue having a carnosinase inhibitor activity,
12. a manganese ion and
13. a zinc ion, or a synergistic combination thereof, in an amount and composition thereof effective to stabilize L-carnosine and its said related substance from enzymatic hydrolysis in the body under preservation of a residual carnosinase activity.
Our own studies of the effects of L-carnosine on human bone marrow stromal cell proliferation supportingly demonstrate that L-carnosine increases a proliferative potential of the human bone marrow cells up to three population doublings, the phenomenon that can be further exploited in rejuvenation and resuscitation cellular and tissue engineering studies (Figure 8). It was shown that intake of carnosine in a dose of 50-100 mg/kg of body weight before X-ray irradiation resulted in an increase of the survival of experimental mice. The protective effect of carnosine was manifested, when it was injected either before or after irradiation, but the effect was more pronounced in the case of shortening time between irradiation and injection. An enhancement of colony forming index of bound cells in spleen was also observed simultaneously with protective action of carnosine [137].
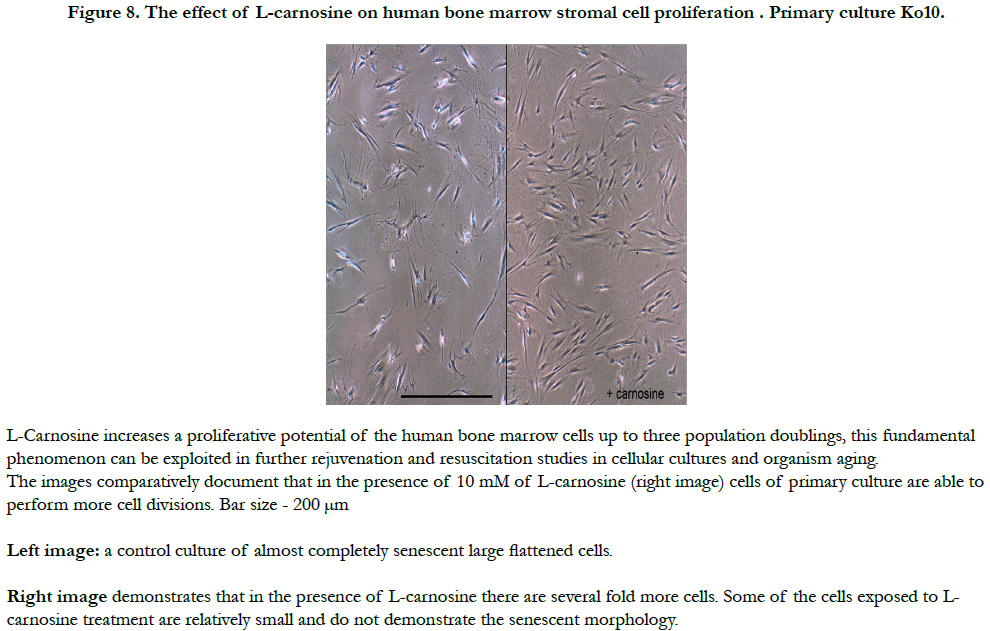
Figure 8. The effect of L-carnosine on human bone marrow stromal cell proliferation . Primary culture Ko10.
Telomere shortening has been implicated in cellular senescence, which may cause certain aging phenotypes. To reveal whether telomere shortening is associated with chronological aging and/ or photoaging of the skin, Sugimoto et al.,[138] have measured telomere length in the epidermis and in the dermis from sun-protected and from sun exposed sites of the skin. Seventy-six specimens of epidermis from sun protected sites and 24 specimens of epidermis from sun exposed sites were analyzed. Sixty specimens of the dermis were also analyzed. In six cases, epidermal specimens from sun-protected and from sun-exposed sites of the same individual were analyzed. Comparison of telomere lengths revealed that the epidermis has shorter telomeres than the dermis. Telomere length in the epidermis and in the dermis was reduced with age, and average telomere shortening rates in the epidermis and in the dermis were 9 and 11 bp/yr, respectively.
Unexpectedly, telomere length was not significantly different between epidermis from sun-exposed sites and from sun-protected sites. The authors could not show the evidence that telomere shortening is associated with photoaging of the skin [138]. Recent work has substantially elucidated the mechanisms of skin aging and photoaging [139]. In particular, a central role for telomere- based signaling can be inferred. Intrinsic aging is largely controlled by progressive telomere shortening, compounded by low grade oxidative damage to telomeres and other cellular constituents, the consequence of aerobic cellular metabolism. In sun exposed skin, UV irradiation also damages DNA and accelerates telomere shortening. Aging and photodamage appear to share a common final pathway that involves signaling through p53 following disruption of the telomere. These telomere-initiated responses, in combination with UV-induced damage to critical regulatory genes, lead to the familiar picture of "photoaging." [139].
Skin is a complex tissue composed of two very different compartments. The continuously renewing epidermis made up mostly by keratinocytes and the underlying matrix-rich dermis with the resting fibroblasts as its major cellular components. Both compartments are tightly interconnected and a paracrine mutual interaction is essential for epidermal growth, differentiation, and tissue homeostasis [140]. Skin aging is commonly viewed as wrinkle formation, hair greying, and impaired wound healing. Nevertheless, the epidermis as the outermost shield needs to remain intact in order to guarantee an inside-out and outside-in barrier function throughout life time of a human being. Furthermore, the epidermis is one of the few regenerative tissues that express telomerase, the ribonucleoprotein complex that can counteract telomere erosion, one of the presently mostly favoured potential mechanisms causing cellular aging. This raises the question whether in the epidermis telomerase is able to counteract telomere erosion and thereby to prevents a telomere-dependent aging process and consequently which part of the skin is responsible for the most obvious changes associated with skin aging [140]. Krunic et al., have demonstrated [141] that telomerase is expressed in the epidermis in situ independent of age but was repressed upon the passaging of keratinocytes in monolayer culture. However, when keratinocytes were grown in organotypic cultures (OTCs), telomerase was re-established, indicating that telomerase activity is not merely proliferation-associated but is regulated in a tissue context-dependent manner in human keratinocytes. While not inducible by growth factors, treatment with the histone deacetylation inhibitor FK228 restored telomerase activity in keratinocytes grown in monolayer cultures. Accordingly, CHIP analyses demonstrated an acetylated, active hTERT promoter in the epidermis in situ and in the epidermis of OTCs but a deacetylated, silenced hTERT promoter with subsequent propagation in monolayer culture suggesting that histone acetylation is part of the regulatory program to guarantee hTERT expression/telomerase activity in the epidermis. In agreement with the loss of telomerase activity, telomeres shortened during continuous propagation in monolayer culture by an average of approximately 70 base pairs (bp) per population doubling (pd). However, telomere erosion varied strongly between different keratinocyte strains and even between individual cells within the same culture, thereby arguing against a defined rate of telomere loss per replication cycle. In the epidermis in situ, as determined from early-passage keratinocytes and tissue sections from different age donors, the authors calculated a telomere loss of only approximately 25 bp per year [141]. Since Krunic et al., determined the same rate for the non-regenerating melanocytes and dermal fibroblasts, their data suggest that in human epidermis telomerase is a protective mechanism against excessive telomere loss during the life-long regeneration [141]. The reason for the sustained expression of telomerase activity in the epidermis is that the formation of a well structured epidermis strictly depends on a tight balance between proliferation and differentiation [142]. Accordingly, telomerase, which is restricted to proliferating cells, is downregulated with differentiation. It is unclear, however, whether this inhibition is essential to or only a consequence of the differentiation process. By studying different variants of the HaCaT skin keratinocytes Cerezo et al.,[142] demonstrated that constitutive overexpression of human telomerase reverse transcriptase (hTERT) in HaCaT-TERT cells (lacking its own differentiation-sensitive promoter) and constitutive expression of the c-myc gene in HaCaT-myc cells caused increased proliferation in conventional cultures; however, this proliferative advantage was not maintained in tissue-like organotypic cocultures. Despite reduced stratification, HaCaT-myc cells were still able to develop a fully differentiated epithelium. HaCaT-TERT cultures, on the other hand, expressed all markers of early but not of terminal differentiation. The failure to differentiate terminally was observed in hTERT mass cultures and individual clones and correlated with an intense nuclear hTERT staining of the uppermost cells of the HaCaT-TERT epithelia. Thus, the published data suggest that constitutive overexpression of hTERT does not interfere with epidermal differentiation per se but blocks the terminal stage of differentiation and therefore indicates that hTERT/telomerase plays an active part in the regulatory pathway of epidermal differentiation [142].
Most human cells lack sufficient telomerase to maintain telomeres, hence these genetic elements shorten with time and stress, contributing to aging and disease. In January, 2007, a commercial health maintenance program, PattonProtocol-1, was launched that included a natural product-derived telomerase activator (TA- 65((R)), 10-50 mg daily), a comprehensive dietary supplement pack, and physician counseling/laboratory tests at baseline and every 3-6 months thereafter [143]. We report here analysis of the first years of data focusing on the protection of telomeres in fibroblasts system using imidazole-containing dipeptides.
In the recent work, the authors studied the effect of carnosine on a human fetal lung fibroblast strain (HPF), which was either kept in a continuously proliferating or proliferation-inhibited state. The results indicate that carnosine can reduce telomere shortening rate possibly by protecting telomere from damage [144]. Based on our own studies, we have suggested that the reduction in telomere shortening rate and damages in telomeric DNA made an important contribution to the life-extension effect of carnosine and carcinine [42, 43].
Cumulatively, in this article we propose that patented specific oral formulations of non-hydrolized carnosine and carcinine provide a powerful tool for targeted inhibition of cumulative oxidative stress and inflammation associated with telomere shortening and senescent phenotype of skin fibroblasts in skin aging and age-related diseases and during smoking behavior (Figures 7,8 ) [42-44, 89]. This product concept implements the potential role of the new nutritional strategy on redox balance in age-related eye diseases and detail how the synergism and interaction of imidazole- containing amino acid-based compounds (nonhydrolized L-carnosine, histidine), chaperone agents (such as, L-carnosine, D-pantethine), glutathione-boosting agents (N-acetylcysteine, vitamin E, methionine), and plays key roles in the function and maintenance of the redox systems in the skin fibroblasts and tissues during aging and in the treatment of human skin disease.
The recently published study investigates whether an orally active non-hydrolized forms of carnosine or carcinine have skin protective and beautification properties in cases of skin aging and clinical diabetic skin manifestations. Treatment with non-hydrolized forms of carnosine and carcinine of patients was accompanied by improvement in skin appearance through the initially proposed transglycation and universal antioxidant mechanisms of action [126]. In the present study, more importantly, the oral formulation of non-hydrolized carnosine is proposed to ameliorate the telomere and telomerase dysfunctions, including telomeres-mediated effects in skin fibroblasts with senescent phenotype of the adult patients. Pharmacological manipulation of those signaling omponents may lead to the prevention and treatment of skin aging and cancer induced by chronic solar exposure. Telomere shortening has not only been linked to aging but also to several age associated diseases, including tumorigenesis, coronary arterydisease, and heart failure. In the current review, we discuss the role of telomere biology in relation to skin aging and aging associated diseases.
Thus, the orally active cellular system of imidazole-containing dipeptide based compounds (non-hydrolized carnosine, carcinine) protect from telomere attrition and are acting as telomerase enhancer as a part of the health maintenance anti-aging program, provide a new option for the clinical treatment of intrinsic skin aging, photoaging and skin diabetic lesions [42-44, 89, 126].
Conclusion
Oxidative damage is the primary cause of skin aging. Skin care products are numerous and overwhelming, yet there is certain similarity among different formulations. Moisturizers are ubiquitous and the presence of particular added ingredients usually supports specific marketing claims [145]. It is believed that oxidative stress is caused by an imbalance between the production of reactive oxygen and a biological system's ability to neutralize the reactive intermediates. Oxidative damage occurs because of both intrinsic and extrinsic mechanisms. Together, intrinsic and extrinsic damage are the primary causes of skin aging. The skin uses a series of intrinsic antioxidants to protect itself from free radical damage. Naturally occurring extrinsic antioxidants have also been widely shown to offset and alleviate these changes [146].
Oral dosage forms of non-hydrolized carnosine or carcinine induce cellular responses in human skin fibroblasts through the telomere-mediated pathway and redox signaling, supporting the view that carnosine or related imidazole-containing dipeptide based compound-induced hormetic stimulation of cellular antioxidant defenses can be a useful approach toward anti-aging intervention to the skin. The present study extends our previous findings and demonstrates that oral application of non-hydrolized forms of carnosine or carcinine in human volunteers significantly increases the antioxidative potential of skin biosurface, thus highlighting the effectiveness of a natural antioxidant biotechnology in the anti-aging management of skin.
Acknowledgements
This work was planned, organized, and supported by Innovative Vision Products, Inc. (County of New Castle, DE, USA). Innovative Vision Products (IVP) is a holder of the worldwide Patents: (WO 2004/028536 A1; WO 94/19325; WO 95/12581; WO 2004/064866 A1) (including most relevant to the article issues PCT International Publication Number WO 2004/064866 PCT/ JP2004/000351) protecting the therapeutic applications and formulations thereof of carnosine and carnosine derivatives stabilizing carnosine from enzymatic hydrolysis. Innovative Vision Products Inc. is a pharmaceutical and Nanotechnology Development Company with a focus on innovative chemical entities, drug delivery systems, and unique medical devices to target specific biomedical applications. Over the last decade IVP has developed a track record in developing these technologies to effectively address the unmet needs of specific diseased populations.
References
- Rabe JH, Mamelak AJ, McElgunn PJ, Morison WL, Sauder DN (2006) Photoaging: mechanisms and repair. J Am Acad Dermatol. 55(1): 1-19.
- Gilchrest BA (2003) Skin aging 2003: recent advances and current concepts. Cutis. 72(3): 5 -10.
- Puizina-Ivić N (2008) Skin aging. Acta Dermatovenerol Alp Panonica Adriat. 17(2): 47-54.
- McCullough JL, Kelly KM (2006) Prevention and treatment of skin aging. Ann N Y Acad Sci. 1067: 323-31.
- Yaar M, Gilchrest B (2008) Aging of skin. Fitzpatrick’s dermatology in general medicine. 7th (edn), New York: McGraw-Hill. 963-73.
- Limpiangkanan W, Limpiangkanan W (2010) Photo-aging: a literature review. J Med Assoc Thai. 93(6): 753-7.
- Lachance PA, Nakat Z, Jeong WS (2001) Antioxidants: an integrative approach. Nutrition. 17(10): 835-8.
- Berneburg M, Plettenberg H, Krutmann J (2000) Photoaging of human skin. Photodermatol Photoimmunol Photomed. 16(6): 239-44.
- Campsi J, Dimri GP, Hara E (1996) "Control of Replicative Senescence ". Handbook of the Biology of Aging. (4th edn), New York: Academic Press. 121–149.
- Cristofalo VJ, Pignolo RJ (1993) Replicative senescence of human fibroblast-like cells in culture. Physiol Rev. 73(3): 617-38.
- GP Dimri, X Lee, G Basile, M Acosta, G Scott, et al., (1995) A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A. 92(20): 9363-7.
- Sachs DL, Voorhees JJ (2010) Age-Reversing Drugs and Devices in Dermatology. Clin Pharmacol Ther. 89(1): 34-43.
- Kulozik M, Krieg T (1989) Changes in collagen connective tissue and fibroblasts in aging. Z Hautkr. 64(11): 1003-4, 1007-9.
- Fisher GJ (2005) The pathophysiology of photoaging of the skin. Cutis. 75(2): 5-8.
- Grinnell F (2003) Fibroblast biology in three-dimensional collagen matrices. Trends Cell Biol. 13(5): 264-9.
- Fisher GJ, Varani J, Voorhees JJ (2008) Looking older: fibroblast collapse and therapeutic implications. Arch Dermatol. 144(5): 666-72.
- Fisher GJ, Datta SC, Talwar HS, Wang ZQ, Varani J, et al., (1996) Molecular basis of sun-induced premature skin ageing and retinoid antagonism. Nature. 379(6563): 335-9.
- Fisher GJ, Kang S, Varani J, Bata-Csorgo Z, Wan Y, et al., (2002) Mechanisms of photoaging and chronological skin aging. Arch Dermatol. 138(11): 1462-70.
- Mays PK, McAnulty RJ, Campa JS, Laurent GJ (1995) Age-related alterations in collagen and total protein metabolism determined in cultured rat dermal fibroblasts: age-related trends parallel those observed in rat skin in vivo. Int J Biochem Cell Biol. 27(9): 937-45.
- Péterszegi G, Andrès E, Molinari J, Ravelojaona V, Robert L (2008) Effect of cellular aging on collagen biosynthesis: I. Methodological considerations and pharmacological applications. Arch Gerontol Geriatr. 47(3): 356-67.
- Kang S, Chung JH, Lee JH, Fisher GJ, Wan YS, et al., (2003) Topical Nacetyl cysteine and genistein prevent ultraviolet-light-induced signaling that leads to photoaging in human skin in vivo. J Invest Dermatol. 120(5): 835-41.
- Fisher GJ, Wang ZQ, Datta SC, Varani J, Kang S, et al., (1997) Pathophysiology of premature skin aging induced by ultraviolet light. N Engl J Med. 337(20): 1419-28.
- Weinstock MA, Bingham SF, Cole GW, Eilers D, Naylor MF, et al., (2001) Reliability of counting actinic keratoses before and after brief consensus discussion: the VA topical tretinoin chemoprevention (VATTC) trial. Arch Dermatol. 137(8): 1055-8.
- Tzellos TG, Klagas I, Vahtsevanos K, Triaridis S, Printza A, et al., (2009) Extrinsic ageing in the human skin is associated with alterations in the expression of hyaluronic acid and its metabolizing enzymes. Exp Dermatol. 18(12): 1028-35.
- Donjacour AA, Cunha GR (1991) Stromal regulation of epithelial function. Cancer Treat Res. 53: 335-64.
- Puizina-Ivić N, Mirić L, Carija A, Karlica D, Marasović D (2010) Modern approach to topical treatment of aging skin. Coll Antropol. 34(3): 1145-53.
- Hadshiew IM, Eller MS, Gilchrest BA (2000) Skin aging and photoaging: the role of DNA damage and repair. Am J Contact Dermat. 11(1): 19-25.
- Bonnefoy M, Drai J, Kostka T (2002) Antioxidants to slow aging, facts and perspectives. Presse Med. 31(25): 1174-84.
- Pierce JD, Cackler AB, Arnett MG (2004) Why should you care about free radicals? RN. 67(1): 38-42.
- Baumann L (2002) Cosmetic dermatology: principles and practice. New York: McGraw-Hill. 226.
- Yaar M, Gilchrest BA (2003) Aging of skin. Fitzpatrick’s dermatology in general medicine. vol 2. New York: McGraw-Hill. 1386–98.
- Ohmura H, Oshimura M (1993) Telomere, cellular senescence and transformation. Nippon Rinsho. 51(7): 1899-906.
- Allsopp RC, Vaziri H, Patterson C, Goldstein S, Younglai EV, et al., (1992) Telomere length predicts replicative capacity of human fibroblasts. Proc Natl Acad Sci U S A. 89(21): 10114-8.
- Oeseburg H, de Boer RA, van Gilst WH, van der Harst P (2010) Telomere biology in healthy aging and disease. Pflugers Arch. 459(2): 259-68.
- Engelhardt M, Martens UM (1998) The implication of telomerase activity and telomere stability for replicative aging and cellular immortality (Review). Oncol Rep. 5(5): 1043-52.
- Aubert G, Lansdorp PM (2008) Telomeres and aging. Physiol Rev. 88(2): 557-79.
- Allsopp RC, Harley CB (1995) Evidence for a critical telomere length in senescent human fibroblasts. Exp Cell Res. 219(1): 130-6.
- Martens UM, Chavez EA, Poon SS, Schmoor C, Lansdorp PM (2000) Accumulation of short telomeres in human fibroblasts prior to replicative senescence. Exp Cell Res. 256(1): 291-9.
- Shammas MA (2010) Telomeres, lifestyle, cancer, and aging. Curr Opin Clin Nutr Metab Care. 14(1): 28-34.
- Babizhayev MA, Meguro K (2004) Combined use of carnosinase inhibitor With I-carsnosines and composition. WO 2004/064866 , PCT/JP2004/000351. Filed 20 January 2003 (20.01.2003).
- Babizhayev MA (2010) New concept in nutrition for the maintenance of the aging eye redox regulation and therapeutic treatment of cataract disease; synergism of natural antioxidant imidazole-containing amino acid-based compounds, chaperone, and glutathione boosting agents: a systemic perspective on aging and longevity emerged from studies in humans. Am J Ther. 17(4): 373-89. https://www.ncbi.nlm.nih.gov/pubmed/20463577
- Babizhayev MA, Yegorov YE (2010) Smoking and health: association between telomere length and factors impacting on human disease, quality of life and life span in a large population-based cohort under the effect of smoking duration. Fundam Clin Pharmacol. 25(4): 425-442.
- Babizhayev MA, Savel’yeva EL, Moskvina SN, Yegorov YE (2010) Telomere Length is a Biomarker of Cumulative Oxidative Stress, Biologic Age, and an Independent Predictor of Survival and Therapeutic Treatment Requirement Associated With Smoking Behavior. Am J Ther. 18(6): e209-226.
- Babizhayev MA, Yegorov YE (2010) Advanced drug delivery of N-acetylcarnosine (N-acetyl-beta-alanyl-L-histidine), carcinine (beta-alanylhistamine) and L-carnosine (beta-alanyl-L-histidine) in targeting peptide compounds as pharmacological chaperones for use in tissue engineering, human disease management and therapy: from in vitro to the clinic. Recent Pat Drug Deliv Formul. 4(3): 198-230.
- Glogau RG (2003) Systemic evaluation of the aging face. Dermatology. Edinburgh: Mosby. 2357–60.
- Wespes E, Schulman CC (2002) Male andropause: myth, reality, and treatment. Int J Impot Res. 14(1): S93-8.
- Phillips TJ, Demircay Z, Sahu M (2001) Hormonal effects on skin aging. Clin Geriatr Med. 17(4): 661-72, vi.
- Arlt W, Hewison M (2004) Hormones and immune function: implications of aging. Aging Cell. 3(4): 209-16.
- Swift ME, Burns AL, Gray KL, DiPietro LA (2001) Age-related alterations in the inflammatory response to dermal injury. J Invest Dermatol. 117(5): 1027-35.
- Arvat E, Broglio F, Ghigo E (2000) Insulin-Like growth factor I: implications in aging. Drugs Aging. 16(1): 29-40.
- Frippiat C, Chen QM, Zdanov S, Magalhaes JP, Remacle J, et al., (2001) Subcytotoxic H2O2 stress triggers a release of transforming growth factorbeta 1, which induces biomarkers of cellular senescence of human diploid fibroblasts. J Biol Chem. 276(4): 2531-7.
- Toussaint O, Medrano EE, von Zglinicki T (2000) Cellular and molecular mechanisms of stress-induced premature senescence (SIPS) of human diploid fibroblasts and melanocytes. Exp Gerontol. 35(8): 927-45.
- Mocali A, Caldini R, Chevanne M, Paoletti F (1995) Induction, effects, and quantification of sublethal oxidative stress by hydrogen peroxide on cultured human fibroblasts. Exp Cell Res. 216(2): 388-95.
- Chen Q, Ames BN (1994) Senescence-like growth arrest induced by hydrogen peroxide in human diploid fibroblast F65 cells. Proc Natl Acad Sci U S A. 91(10): 4130-4.
- Toussaint O, Houbion A, Remacle J (1992) Aging as a multi-step process characterized by a lowering of entropy production leading the cell to a sequence of defined stages. II. Testing some predictions on aging human fibroblasts in culture. Mech Ageing Dev. 65(1): 65-83.
- von Zglinicki T, Saretzki G, Döcke W, Lotze C (1995) Mild hyperoxia shortens telomeres and inhibits proliferation of fibroblasts: a model for senescence? Exp Cell Res. 220(1): 186-93.
- Bayreuther K, Rodemann HP, Hommel R, Dittmann K, Albiez M, et al., (1988) Human skin fibroblasts in vitro differentiate along a terminal cell lineage. Proc Natl Acad Sci U S A. 85(14): 5112-6.
- Bumann J, Santo-Höltje L, Löffler H, Bamberg M, Rodemann HP (1995) Radiation-induced alterations of the proliferation dynamics of human skin fibroblasts after repeated irradiation in the subtherapeutic dose range. Strahlenther Onkol. 171(1): 35-41.
- Dumont P, Burton M, Chen QM, Gonos ES, Frippiat C, et al., (2000) Induction of replicative senescence biomarkers by sublethal oxidative stresses in normal human fibroblast. Free Radic Biol Med. 28(3): 361-73.
- Lavker RM, Zheng PS, Dong G (1987) Aged skin: a study by light, transmission electron, and scanning electron microscopy. J Invest Dermatol. 88(3): 44s-51s.
- Tobin DJ, Paus R (2001) Graying: gerontobiology of the hair follicle pigmentary unit. Exp Gerontol. 36(1): 29-54.
- Piérard GE, Uhoda I, Piérard-Franchimont C (2003) From skin microrelief to wrinkles. An area ripe for investigation. J Cosmet Dermatol. 2(1): 21-8.
- Dayan D, Abrahami I, Buchner A, Gorsky M, Chimovitz N (1988) Lipid pigment (lipofuscin) in human perioral muscles with aging. Exp Gerontol. 23(2): 97-102.
- Donofrio LM (2000) Fat distribution: a morphologic study of the aging face. Dermatol Surg. 26(12): 1107-12.
- Butterwick KJ, Lack EA (2003) Facial volume restoration with the fat autograft muscle injection technique. Dermatol Surg. 29(10): 1019-26.
- Ramirez OM, Robertson KM (2001) Comprehensive approach to rejuvenation of the neck. Facial Plast Surg. 17(2): 129-40.
- Bolognia JL, Braverman IM, Rousseau ME, Sarrel PM (1989) Skin changes in menopause. Maturitas. 11(4): 295-304.
- Scharffetter-Kochanek K, Brenneisen P, Wenk J, Herrmann G, Ma W, et al., (2000) Photoaging of the skin from phenotype to mechanisms. Exp Gerontol. 35(3): 307-16.
- Braverman IM, Fonferko E (1982) Studies in cutaneous aging: I. The elastic fiber network. J Invest Dermatol. 78(5): 434-43.
- Talwar HS, Griffiths CE, Fisher GJ, Hamilton TA, Voorhees JJ (1995) Reduced type I and type III procollagens in photodamaged adult human skin. J Invest Dermatol. 105(2): 285-90.
- Varani J, Spearman D, Perone P, Fligiel SE, Datta SC, et al.,(2001) Inhibition of type I procollagen synthesis by damaged collagen in photoaged skin and by collagenase-degraded collagen in vitro. Am J Pathol. 158(3): 931-42.
- Ferguson J, Dover JS (2006) Photodermatology. London: Manson Publishing. 160.
- Sander CS, Chang H, Salzmann S, Müller CS, Ekanayake-Mudiyanselage S, et al., (2002) Photoaging is associated with protein oxidation in human skin in vivo. J Invest Dermatol. 118(4): 618-25.
- Fisher GJ, Talwar HS, Lin J, Lin P, McPhillips F, et al., (1998) Retinoic acid inhibits induction of c-Jun protein by ultraviolet radiation that occurs subsequent to activation of mitogen-activated protein kinase pathways in human skin in vivo. J Clin Invest. 101(6): 1432-40.
- Sams WM Jr (1986) Sun-induced aging. Clinical and laboratory observations in man. Dermatol Clin. 4(3): 509-16.
- Sørensen LT, Jørgensen S, Petersen LJ, Hemmingsen U, Bülow J, et al., (2008) Acute effects of nicotine and smoking on blood flow, tissue oxygen, and aerobe metabolism of the skin and subcutis. J Surg Res. 152(2): 224-30.
- Thomsen SF, Sørensen LT (2010) Smoking and skin disease. Skin Therapy Lett. 15(6): 4-7.
- Smith JB, Fenske NA (1996) Cutaneous manifestations and consequences of smoking. J Am Acad Dermatol. 34(5 pt 1): 717-32.
- Sørensen LT, Nielsen HB, Kharazmi A, Gottrup F (2004) Effect of smoking and abstention on oxidative burst and reactivity of neutrophils and monocytes. Surgery. 136(5): 1047-53.
- Sørensen LT, Zillmer R, Agren M, Ladelund S, Karlsmark T, et al., (2009) Effect of smoking, abstention, and nicotine patch on epidermal healing and collagenase in skin transudate. Wound Repair Regen. 17(3): 347-53.
- Jorgensen LN, Kallehave F, Christensen E, Siana JE, Gottrup F (1998) Less collagen production in smokers. Surgery. 123(4): 450-5.
- Sørensen LT, Hemmingsen U, Kallehave F, Wille-Jørgensen P, Kjaergaard J, et al., (2005) Risk factors for tissue and wound complications in gastrointestinal surgery. Ann Surg. 241(4): 654-8.
- Demierre MF, Brooks D, Koh HK, Geller AC (1999) Public knowledge, awareness, and perceptions of the association between skin aging and smokking. J Am Acad Dermatol. 41(1): 27-30.
- Grady D, Ernster V (1992) Does cigarette smoking make you ugly and old? Am J Epidemiol. 135(8): 839-42.
- Lahmann C, Bergemann J, Harrison G, Young AR (2001) Matrix metalloproteinase- 1 and skin ageing in smokers. Lancet. 357(9260): 935-6.
- Wenk J, Brenneisen P, Meewes C, Wlaschek M, Peters T, et al., (2001) UVinduced oxidative stress and photoaging. Curr Probl Dermatol. Oxidants and Antioxidations in cutaneous biology. 29: 83-94.
- Perricone N (2001) The wrinkle cure: Unlock the power of Cosmeceuticals for Supple, Youthful skin. New York: Warner Books. 207.
- Boldyrev AA, Severin SE (1990 ) The histidine-containing dipeptides, carnosine and anserine: distribution, properties and biological significance. Adv Enzyme Regul. 30: 175-194.
- Babizhayev MA, Yegorov YE (2010) Therapeutic uses of drug-carrier systems for imidazole-containing dipeptide compounds that act as pharmacological chaperones and have significant impact on the treatment of chronic diseases associated with increased oxidative stress and the formation of advanced glycation end products. Crit Rev Ther Drug Carrier Syst. 27(2): 85-154.
- Lenney JF (1976) Specificity and distribution of mammalian carnosinase. Biochim Biophys Acta. 429(1): 214-9.
- Jackson MC, Kucera CM, Lenney JF (1991) Purification and properties of human serum carnosinase. Clin Chim Acta. 196(2-3): 193-205.
- Lenney JF (1990) Separation and characterization of two carnosine-splitting cytosolic dipeptidases from hog kidney (carnosinase and non-specific dipeptidase). Biol Chem Hoppe Seyler. 371(5): 433-40.
- Kunze N, Kleinkauf H, Bauer K (1986) Characterization of two carnosinedegrading enzymes from rat brain. Partial purification and characterization of a carnosinase and a beta-alanyl-arginine hydrolase. Eur J Biochem. 160(3): 605-13.
- Aldini G, Facino RM, Beretta G, Carini M (2005) Carnosine and related dipeptides as quenchers of reactive carbonyl species: from structural studies to therapeutic perspectives. Biofactors. 24(1-4): 77-87.
- Aldini G, Orioli M, Carini M, Maffei Facino R (2004) Profiling histidinecontaining dipeptides in rat tissues by liquid chromatography/electrospray ionization tandem mass spectrometry. J Mass Spectrom. 39(12): 1417-28.
- Reddy VP, Garrett MR, Perry G, Smith MA (2005) Carnosine: a versatile antioxidant and antiglycating agent. Sci Aging Knowledge Environ. 2005(18): pe12.
- Rashid I, van Reyk DM, Davies MJ (2007) Carnosine and its constituents inhibit glycationof low-density lipoproteins that promotes foam cell formation in vitro. FEBS Lett. 581(5): 1067–70.
- Babizhayev MA (1989) Antioxidant activity of L-carnosine, a natural histidine-containing dipeptide in crystalline lens. Biochim Biophys Acta. 1004(3): 363-71.
- Babizhayev MA, Micans P, Guiotto A, Kasus-Jacobi A (2009) N-acetylcarnosine lubricant eyedrops possess all-in-one universal antioxidant protective effects of L-carnosine in aqueous and lipid membrane environments, aldehyde scavenging, and transglycation activities inherent to cataracts: a clinical study of the new vision-saving drug N-acetylcarnosine eyedrop therapy in a database population of over 50,500 patients. Am J Ther. 16(6): 517-33.
- Babizhayev MA (2009) Current ocular drug delivery challenges for N-acetylcarnosine: novel patented routes and modes of delivery, design for enhancement of therapeutic activity and drug delivery relationships. Recent Pat Drug Deliv Formul. 3(3): 229-65.
- Babizhayev MA, Kasus-Jacobi A (2009) State of the art clinical efficacy and safety evaluation of N-acetylcarnosine dipeptide ophthalmic prodrug. Principles for the delivery, self-bioactivation, molecular targets and interaction with a highly evolved histidyl-hydrazide structure in the treatment and therapeutic management of a group of sight-threatening eye diseases. Curr Clin Pharmacol. 4(1): 4-37.
- Babizhayev MA, Deyev AI, Yermakova VN, Semiletov YA, Davydova NG, et al., (2001) N-Acetylcarnosine, a natural histidine-containing dipeptide, as a potent ophthalmic drug in treatment of human cataracts. Peptides. 22(6): 979-994.
- Babizhayev MA, Deyev AI, Yermakova VN, Semiletov YA, Davydova NG, et al., Efficacy of N-acetylcarnosine in the treatment of cataracts. Drugs RD. 3(2): 87-103.
- Babizhayev MA, Deyev AI, Yermakova VN, Remenschikov VV, Bours J (2006) Revival of the lens transparency with N-acetylcarnosine. Current Drug Therapy. 1: 91-116.
- Babizhayev MA, Deyev AI, Yermakova VN, Brikman IV, Bours J et al., (2004) Lipid peroxidation and cataracts: N-acetylcarnosine as a therapeutic tool to manage age-related cataracts in human and in canine eyes. Drugs R D. 5(3): 125-139.
- Babizhayev MA (2004) Method for topical treatment of eye disease and composition and device for said treatment. PCT Patent Application. International Publication Number WO 2004/028536 A1.
- Babizhayev MA (2006) Ophthalmic pharmacology of N-acetylcarnosine lubricant eye drops . J Pharmacol Toxicol. 1(3): 201-233.
- Babizhayev MA (2004) Rejuvenation of visual functions in older adult drivers and drivers with cataract during a short-term administration of N-acetylcarnosine lubricant eye drops. Rejuvenation Res. 7(3): 186-198.
- Babizhayev MA, Yermakova VN, Deyev AI, Seguin MC (2000) Imidazolecontaining peptidomimetic NACA as a potent drug for the medicinal treatment of age-related cataract in humans. J Anti-Aging Medicine. 3(1): 43-62.
- Arnould JM, Frentz R (1975) Presence, isolation and chemical structure of a substance characteristic of cardiac tissue in Carcinus maenas (L.): betaalanylhistamine. Comp Biochem Physiol C. 50(1): 59-66.
- Brotman DN, Flancbaum L, Fitzpatrick JC, Fisher H (1989) Presence of carcinine (β-alanylhistamine) in mammalian tissues. FASEB J. 3: 1028.
- Brotman DN, Flancbaum L, Kang YH, Merrill GF, Fisher H (1990) Positive inotropic effects of carcinine in the isolated, perfused guinea pig heart. Crit Care Med. 18(3): 317–321.
- Flancbaum L, Brotman DN, Fitzpatrick JC, Van Es T, Kasziba E, et al., (1990) Existence of carcinine, a histamine-related compound, in mammalian tissues. Life Sci. 47(17): 1587–1593.
- Ali SM, Tedford CE, Gregory R, Handley MK, Yates SL, et al., (1999) Design, synthesis, and structure–activity relationships of acetylene-based histamine H3 receptor antagonists. J Med Chem. 42(5): 903–909.
- Tedford CE, Phillips JG, Gregory R, Pawlowski GP, Fadnis L, et al., (1999) Development of trans-2-[1H-imidazol-4-yl] cyclopropane derivatives as new high-affinity histamine H3 receptor ligands. J Pharmacol Exp Ther. 289(2): 1160–1168.
- Yates SL, Phillips JG, Gregory R, Pawlowski GP, Fadnis L, et al., (1999) Identification and pharmacological characterization of a series of new 1H-4 substituted-imidazoyl histamine H3 receptor ligands. J Pharmacol Exp Ther. 289(2): 1151-9.
- Tozer MJ, Kalindjian SB (2000) Histamine H3 receptor antagonists. Expert Opin Ther. Patents. 10(7): 1045–1055.
- Chen Z, Sakurai E, Hu W, Jin C, Kiso Y, et al., (2004) Pharmacological effects of carcinine on histaminergic neurons in the brain. Br J Pharmacol. 143(5): 573-80.
- Babizhayev MA (2006) Biological activities of the natural imidazole-containing peptidomimetics n-acetylcarnosine, carcinine and L-carnosine in ophthalmic and skin care products. Life Sci. 78(20): 2343-57. https://www. ncbi.nlm.nih.gov/pubmed/16388826
- Babizhayev MA, Seguin MC, Gueyne J, Evstigneeva RP, Ageyeva EA, et al., (1994) L-carnosine (beta-alanyl-L-histidine) and carcinine (beta-alanylhistamine) act as natural antioxidants with hydroxyl-radical-scavenging and lipid-peroxidase activities. Biochem J. 304 (2): 509-16.
- Arnould JM, Frentz R (1977) Carcinine (β-alanyl-histamine): rapid synthesis and action on vertebrate blood pressure. Arch Int Physiol Biochim. 85(2): 339-50.
- Babizhayev MA, Kasus-Jacobi A (2009) Imidazole-containing peptidebased compounds in sight-threatening age-related eye diseases. Expert Rev Ophthalmol. 4(6): 581–586.
- Babizhayev MA (2010) Designation of imidazole-containing dipeptides as pharmacological chaperones. Hum Exp Toxicol. 30(7): 736-761.
- Babizhayev MA, Nikolayev GM, Nikolayeva JG, Yegorov YE (2010) Biologic Activities of Molecular Chaperones and Pharmacologic Chaperone Imidazole-Containing Dipeptide-Based Compounds: Natural Skin Care Help and the Ultimate Challenge. Implication for Adaptive Responses in the Skin. Am J Ther. 19(2): e69-89.
- Babizhayev MA, Deyev AI (2012) Management of the Virulent Influenza Virus Infection by Oral Formulation of Nonhydrolized Carnosine and Isopeptide of Carnosine Attenuating Proinflammatory Cytokine-Induced Nitric Oxide Production. Am J Ther. 19(1) : e25-47.
- Babizhayev MA, Deyev AI, Savel’yeva EL, Lankin VZ, Yegorov YE (2011) Skin beautification with oral non-hydrolized versions of carnosine and carcinine: Effective therapeutic management and cosmetic skincare solutions against oxidative glycation and free-radical production as a causal mechanism of diabetic complications and skin aging. J Dermatol Treat. 23(5): 345-81.
- Holliday R, McFarland GA (2000) A role for carnosine in cellular maintenance. Biochemistry (Mosc). 65(7): 843–848.
- McFarland GA, Holliday R (1994) Retardation of the senescence of cultured human diploid fibroblasts by carnosine. Exp Cell Res. 212(2): 167–175.
- McFarland GA, Holliday R (1999) Further evidence for the rejuvenating effects of the dipeptide L-carnosine on cultured human diploid fibroblasts. Exp Gerontol. 34(1): 35–45.
- Peterszegi G, Molinari J, Ravelojaona V, Robert L (2006) Effect of advanced glycation end-products on cell proliferation and cell death. Pathol Biol (Paris). 54(7): 396-404.
- Hipkiss AR (2010) Aging, Proteotoxicity, Mitochondria, Glycation, NAD+ and Carnosine: Possible Inter-Relationships and Resolution of the Oxygen Paradox. Front Aging Neurosci. 2: 10.
- Hipkiss AR, Preston JE, Himsworth DT, Worthington VC, Keown M, et al., (1998) Pluripotent protective effects of carnosine, a naturally occurring dipeptide. Ann N Y Acad Sci. 854: 37-53.
- Renner C, Zemitzsch N, Fuchs B, Geiger KD, Hermes M, et al., (2010) Carnosine retards tumor growth in vivo in an NIH3T3-HER2/neu mouse model. Mol Cancer. 9: 2.
- Ikeda D, Wada S, Yoneda C, Abe H, Watabe S (1999) Carnosine stimulates vimentin expression in cultured rat fibroblasts. Cell Struct Funct. 24(2): 79-87.
- Kantha SS, Wada S, Tanaka H, Takeuchi M, Watabe S, et al., (1996) Carnosine sustains the retention of cell morphology in continuous fibroblast culture subjected to nutritional insult. Biochem Biophys Res Commun. 223(2): 278-82.
- Oya Y, Yamamoto K (1988) The biological activity of hydrogen peroxide. IV. Enhancement of its clastogenic actions by coadministration of L-histidine. Mutat Res. 198(1): 233-40.
- Kurella EG, Mal'tseva VV, Seslavina LS, Stvolinskiĭ SL (1991) Stimulating effects of carnosine on hemopoietic stem cells. Biull Eksp Biol Med. 112(7): 52-3.
- Sugimoto M, Yamashita R, Ueda M (2006) Telomere length of the skin in association with chronological aging and photoaging. J Dermatol Sci. 43(1): 43-7.
- Kosmadaki MG, Gilchrest BA (2004) The role of telomeres in skin aging/photoaging. Micron. 35(3): 155-9.
- Boukamp P (2005) Skin aging: a role for telomerase and telomere dynamics? Curr Mol Med. 5(2): 171-7.
- Krunic D, Moshir S, Greulich-Bode KM, Figueroa R, Cerezo A, et al., (2009) Tissue context-activated telomerase in human epidermis correlates with little age-dependent telomere loss. Biochim Biophys Acta. 1792(4): 297-308.
- Cerezo A, Stark HJ, Moshir S, Boukamp P (2003) Constitutive overexpression of human telomerase reverse transcriptase but not c-myc blocks terminal differentiation in human HaCaT skin keratinocytes. J Invest Dermatol. 121(1): 110-9.
- Harley CB, Liu W, Blasco M, Vera E, Andrews WH, et al., (2010) A Natural Product Telomerase Activator As Part of a Health Maintenance Program. Rejuvenation Res. 14(1): 45-56.
- Shao L, Li QH, Tan Z (2004) L-carnosine reduces telomere damage and shortening rate in cultured normal fibroblasts. Biochem Biophys Res Commun. 324(2): 931-6.
- Cecchi T, Cecchi P, Passamonti P (2010) The first quantitative rating system of the antioxidant capacity of beauty creams via the Briggs-Rauscher reaction: a crucial step towards evidence-based cosmetics. Analyst. 136(3): 613-618.
- Palmer DM, Kitchin JS (2010) Oxidative damage, skin aging, antioxidants and a novel antioxidant rating system. J Drugs Dermatol. 9(1): 11-5.
- Moldaver MV, Yegorov YE (2009) Sparse plating increases the heterogeneity of proliferative potential of fibroblasts. Mech Ageing Dev. 130(5): 337-42.





