Stereoselective Synthesis of C1-C6 and C7-C22 Fragments of (-) Callystatin A
Kurra Yadagiri1, Dasari Madhuri2, Gowravaram Sabitha3, J.S. Yadav4*
1 Department of Chemistry, Texas A&M University, College station, Texas, USA.
2 Department of Chemistry, Emory University, Atlanta, Georgia, USA.
3 Natural products Chemistry Division, CSIR-Indian Institute of Chemical Technology, Hyderabad 500007, India.
4 Centre for Semiochemicals, CSIR-Indian Institute of Chemical Technology, Hyderabad, India.
*Corresponding Author
Dr. J. S. Yadav,
Centre for Semiochemicals,
CSIR-Indian Institute of Chemical Technology,
Hyderabad, India.
E-mail: yadav@iict.res.in
Received: September 05, 2015; Accepted: October 15, 2015; Published: October 21, 2015
Citation: Kurra Yadagiri, Dasari Madhuri, Gowravaram Sabitha, J.S. Yadav (2015) Stereoselective Synthesis of C1-C6 and C7-C22 Fragments of (-) Callystatin A. Int J Bioorg Chem Mol Biol, 3(1), 1-11. doi:dx.doi.org/10.19070/2332-2756-150001
Copyright: J.S. Yadav© 2015. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
Abstract
The stereoselective synthesis of the two major fragments (C1-C6 and C7-C22) of cytotoxic polyketide marine natural product (-) callystatin A, has been achieved with Sharpless epoxidation, desymmetrization strategy, Horner-Wadsworth-Emmons reaction and witting olefination.
2.Introduction
3.Results and Discussion
3.1.Synthetic Strategy for C1–C6 Fragment (2)
3.2.Synthetic Strategy for C7–C12 Fragment (5)
3.3.Synthetic Strategy for C13–C22 Fragment (6)
3.4.Synthesis of C7-C22 fragment (3)
4.Conclusion
5.Experimental Section
5.1.General
5.2.Dimer Compound
6.Acknowledgement
7.References
Keywords
(–)-Callystatin A; Desymmetrization; Horner-Wadsworth-Emmons reaction.
Introduction
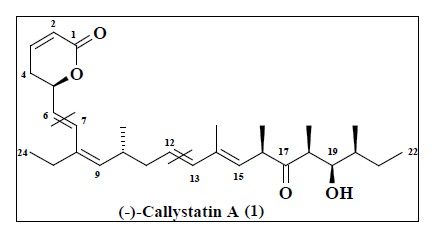
Callystatin A, biologically potent marine natural product with intricate structural features are always attractive synthetic targets to organic chemists. Natural products of marine origin are generally obtained in minute quantities (1 mg from 10 kg of sponge from callyspongia truncate [1]) that are insufficient for detailed biological activity studies. In 1997 Kobayashi and co-workers disclosed the isolation and planar structure of (-) callystatin A [1], a remarkably potent cytotoxic agent (e. g. IC50 0.01 ng/mL in vitro against the KB cancer cell line). Kobayashi group determined the absolute configuration of the (–)-callystatin A via partial [2] and total synthesis [3] by preparing several structural analogues, which led to further insight on structure-activity relationships [4]. The structure of (–)-callystastin A contains a polypropionate chain and a lactone ring connected to each other by two conjugated diene systems separated by two sp3 hybridised carbon atoms.
Interesting structural features combined with the important biological activity of (-) callystatin A has attracted several research groups to attempt its total synthesis [3] as well as the synthesis of its analogues.
Results and Discussion
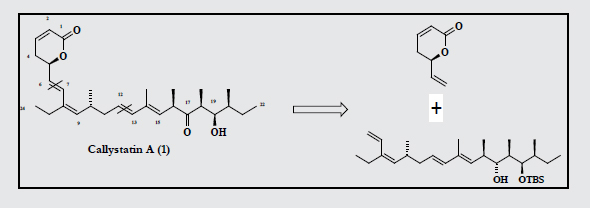
We devised a general retrosynthetic strategy that leads to three building blocks of comparable molecular complexity (Scheme 1).
In this paper, we report scalable synthesis of the C1-C6 fragment 2 from 3-butyne-1-ol, C7-C12 fragment 5 from (S)-Roche ester and C13-C22 fragment 6 from a bicyclic olefin 10 using desymmetrization strategy. We achieved the fragment (C7-C22) using the convergent approach joining the two subunits (5 and 6) together with Wittig olefination. Preliminary results were published recently as a communication [5]. In literature the formation of C13-C22 propionate fragment was to be constructed through convergent synthesis with expensive reagents [6-8]. The formation methyl chiral substrates is very difficult with appropriate configuration. In this study, we have employed the desymmetrization strategy with linear protocol.
The synthesis of the C1-C6 fragment (2) based on sequence of reactions starting from the commercial available compound 3-butyne-1-ol 7. The compound 7 was protected as its p-methoxy benzyl ether using PMBBr and NaH in dry THF at room temperature to afford the compound 11 in 81% yield [9]. The compound 11 was treated with the Grignard reagent prepared from ethyl bromide and magnesium followed by quenching with para-formaldehyde in dry THF afforded compound 12 in 85% yield (Scheme 2). The treatment of 12 with lithium aluminum hydride (LAH) in dry THF at room temperature furnished trans allylic alcohol 13 in 80% yield. The allylic alcohol 13 upon Sharpless Asymmetric epoxidation [10] using (-) DET afforded the corresponding epoxide 14 in 70% yield. The hydroxyl group of 14 was converted into its iodo compound 15 in 81% yield. The compound 14 was converted into a secondary allylic alcohol 4 in 85% yield [11] by refluxing with activated Zinc in dry ethanol. The secondary hydroxyl group 4 was converted into its corresponding methoxy ether 16 using Hung's base and MOMCl in 88% yield [12]. The oxidative removal of the PMB ether with DDQ [13] in CH2Cl2/H2O (9:1) gave alcohol 17 in 75% yield. The primary hydroxyl group of 17 was subjected to oxidation with IBX [14] to yield the compound 18 in 72% yield. The Z-alkene 19 was obtained from aldehyde 18 using Horner-Wadsworth-Emmons Olefination protocol [15]. The compound 19 was treated with catalytic amount of p-TSA in dry benzene results in the formation of C1-C6 fragment 2 in 85% yield (Scheme 2).
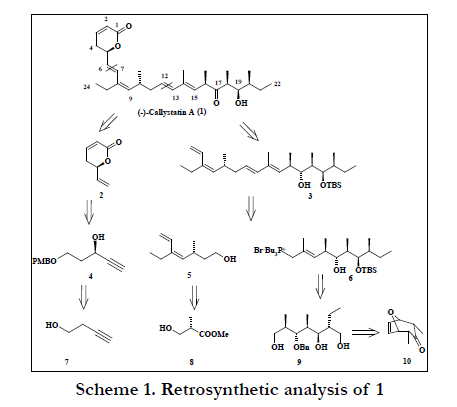
Scheme 1. Retrosynthetic analysis of 1
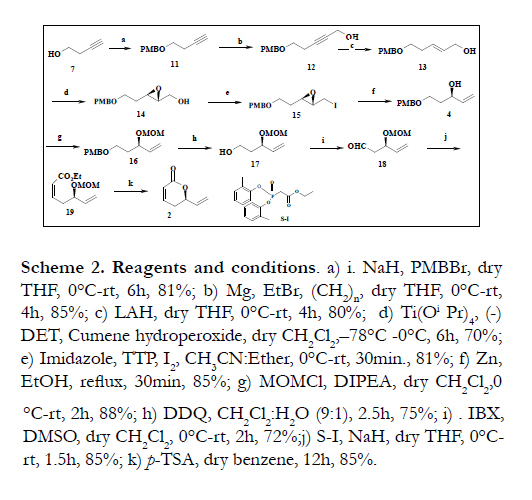
Scheme 2. Reagents and conditions
The construction of the C7-C12 fragment was initiated with the preparation of protected (S)-(+) Roche ester 20 from (S)- (+) Roche ester 8 using PMB imidate and p-TSA in 90% yield (Scheme 3). The reduction of ester 20 was treated with LiBH4 generated in situ) in dry THF to afforded alcohol 21 in 90% yield. This alcohol was oxidized with IBX to give aldehyde 22 in 85% yield and homologated with (methylene) triphenyl phosphorane in dry THF using n-BuLi (1.6 M) to afford alkene 23 in 60% yield [16]. The hydroboration of compound 23 using BH3. Me2S complex in dry THF afforded alcohol 24 in 65% yield and the free hydroxyl group was silylated with TBDPS-Cl to furnish the protected compound 25 in 92% yield. Subsequently the oxidative removal of the PMB ether with DDQ in CH2Cl2/H2O (9:1) gave alcohol 26 in 85% yield. The primary hydroxyl group of 26 was subjected to oxidation with IBX [14] to yield the compound 27 in 82% yield. The Z-alkene 28 was obtained from aldehyde 27 using Horner-Wadsworth-Emmons Olefination protocol [15]. The ester compound 28 was treated with DIBAL-H in anhydrous CH2Cl2 at -78°C to afforded the allyl alcohol 29 in 90% yield. This alcohol was oxidized with IBX to give aldehyde 30 in 80% yield and homologated with (methylene) triphenyl phosphorane in dry THF using n-BuLi (1.6 M) to afford alkene 31 in 65% yield [16] The compound 31 was desilylation with TBAF led to the construction of the C7-C12 fragment 5 in 85% yield (Scheme 3).
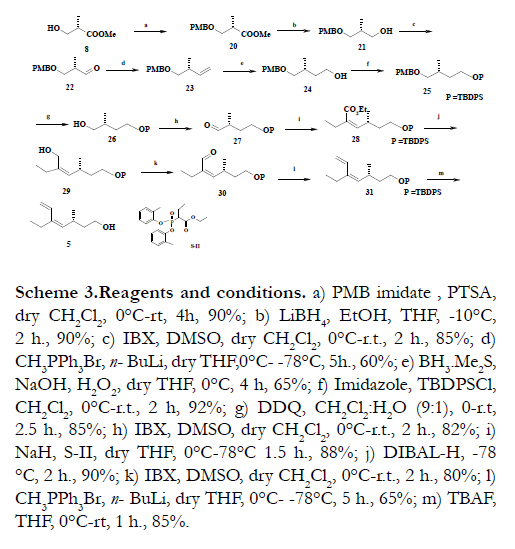
Scheme 3.Reagents and conditions
As depicted in Scheme 4, the construction of the C13-C22 segment was initiated with the preparation of benzyl protected compound 32 from bicyclic alcohol 10 [17]. The bicyclic olefin 32 was subjected to the key desymmetrization reaction using the chiral hydroboration reaction of Brown et al.[18] to afford the required alcohol 33 in 90% yield with good enantio and regioselectivity. The alcohol 33 was oxidized with PCC [19] to furnish the corresponding ketone 34 in 85% yield. The ketone 34 was further oxidized to yield lactone 35 in 90% under Bayer-Villiger conditions [20]. The bicyclic lactone 35 was then subjected to enolization using LDA in THF at -78°C followed by treatment with Ethyl iodide to furnish the ethylated lactone 36 as single diastereomer in 85% yield. Reductive ring-opening of the lactone 36 using excess LiAlH4 resulted in the triol 9 in 80% yield with four chiral centers. The 1, 3-diol functionality in triol 9 was protected as the acetonide using 2, 2-DMP and catalytic amount p-TSA in CH2Cl2 at 0°C to afford 37 in 80% yield and the free hydroxyl group was Pivolylated with Pivolyl chloride to furnish the fully protected triol 38 in 85% yield. Subsequently, the deprotection of acetonide group with p-TSA MeOH led to the diol 40 in 75% yield. The primary hydroxyl group of compound 40 was tosylated with tosyl chloride to afford the compound 41 in 70% yield. The secondary hydroxyl group of 41 was silylated using TBDMSOTf [21] and 2, 6-lutidine to give 42 in 85% yield. The reductive cleavage of pivaloyl group as well as tosyl group in compound 42 with LiAlH4 to yield alcohol 42 in 85% yield. Debenzylation of compound 42 using Li metal and liq NH3 to afford the compound 43 in 80% yield. Primary hydroxyl group of diol compound 43 was selectively oxidized under Swern [22] oxidation conditions using (COCl)2, DMSO and Et3N at –78°C followed by Wittig reaction with carbethoxyethylidene triphenylphosphorane, in refluxing dry CH2Cl2 to give α, β-unsaturated ester 44 in 80% overall yield for the two step sequence (Scheme 4).
The α, β-unsaturated ester 44 on DIBAL-H [23] reduction gave allylic alcohol 45 in 80% yield, which was then converted to allyl bromide 46 using PPh3, 2, 6 lutidine and CBr4 in dry CH3CN in 93% yield. The allylic bromide 46 was converted to its phosphonium salt 6 using PBu3, completing the synthesis of C13-C22 fragment.
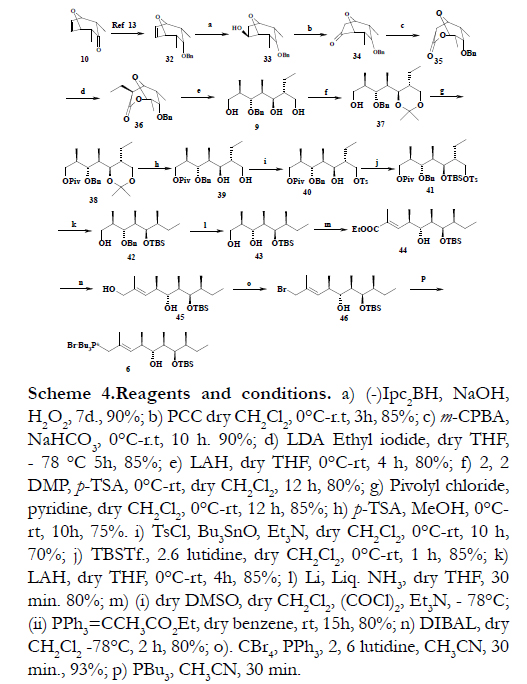
Scheme 4.Reagents and conditions
The primary hydroxyl group of 5 was subjected to oxidation with IBX to yield the compound 47 in 85% yield and the coupling of 47 was done by treatment with phosphonium salt 6 in the presence of LiCH2S(O)CH3 in toluene at -78°C to give C7-C22 fragment 3 in 60% yield (Scheme 5). All that remained to complete the synthesis of (-)-callystatin A (1) is to couple the fragment, 3 with the lactone 2 and followed by functional group manipulations. But, however the cross-metathesis reaction between the diene 3 and vinyl lactone 2 in the presence of Grubb’s-II catalyst failed to give the product, instead the formation of dimer of vinyl lactone 2 was observed as major product. Due to the high reactivity of vinyl lactone with Grubbs's catalyst, it undergoes self condensation rather than a condensation with the other fragment 3.
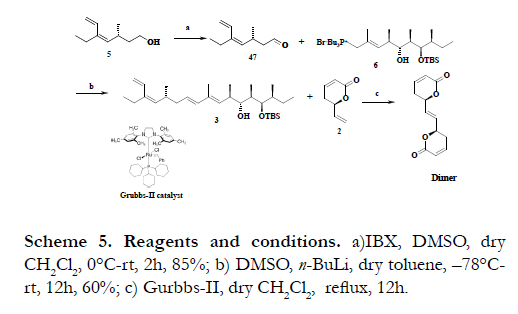
Scheme 5. Reagents and conditions
Conclusion
In conclusion, we have accomplished the C1–C6 and C7–C22 Fragments of (–)- callystatin A in a highly convergent way, by using desymmetrization strategy and Horner-Wadsworth- Emmons reaction.
Experimental Section
All reactions were carried out under an inert atmosphere of argon or nitrogen using standard syringe, septa, and cannula techniques unless otherwise mentioned. Commercial reagents were used without further purification. All solvents were purified by standard techniques. Infrared (IR) spectra were recorded with a Perkin– Elmer 683 spectrometer with NaCl optics. Spectra were calibrated against the Polystyrene absorption at 1610 cm–1. Samples were scanned neat, in KBr wafers or in chloroform as a thin film. 1H NMR spectra were recorded in CDCl3 with a Bruker 300, Varian Unity 500 NMR spectrometer. 13C NMR spectra were recorded at 75MHz in CDCl3 using Tetramethylsilane as the reference standard. Column chromatography was performed using silica gel (60-120 mesh) and the column was usually eluted with ethyl acetate-petroleum ether. Visualization of the spots on TLC plates was achieved either by exposure to iodine vapor or UV light or by dipping the plates to sulphuric acid-β-naphthol or to ethanolic anisaldehyde-sulphuric acid-acetic acid or to phosphomolybdic acid-sulphuric acid solution and heating the plates at 120°C. Mass spectra were obtained on a Finnigan MAT1020B or micromass VG 70-70H spectrometer operating at 70 eV using a direct inlet system. Optical rotations were recorded on high sensitive polarimeter with 10mm cell.
To a stirred suspension of freshly activated NaH (17.14 g, 714.28 mmol) in dry THF (150 mL) under N2 atmosphere was added 7 (20.0 g, 285.70 mmol) in dry THF (50 mL) in a dropwise manner at 0°C. After stirring for 30 min at 0°C, PMB-Br (22.14 g, 285.70 mmol) was added dropwise. The reaction mixture was stirred for 6 h at 0°C, and quenched with saturated KBr solution. The layers were separated and aq. layer was extracted with ethyl acetate (2x100 mL). The combined organic layers were washed with water, brine solution and then dried over anhydrous Na2SO4. Solvent was removed in vacuo and the residue was purified by silica gel column chromatography (EtOAc/pet.ether, 1:9) to afford 11 (43.9 g, 81% yield) as viscous liquid. 1H NMR (CDCl3, 200 MHz): δ 7.21 (d, 2H, J = 8.3 Hz), 6.82 (d, 2H, J = 8.3 Hz), 4.45 (s, 2H), 3.78 (s, 3H), 3.52 (t, 2H, J = 7.5 Hz), 2.44 (dt, 2H, J = 2.5, 7.5 Hz), 1.87 (t, 1H, J = 2.5 Hz).
Freshly prepared EtMgBr (prepared in situ from 8.14 g (339.16 mmol) of Mg and 26.18 mL (339.16 mmol) of ethyl bromide in 60 mL of dry THF) was added dropwise to stirred solution of alkyne 11 (43.0 g, 226.31 mmol) in dry THF (200 mL) at 0°C. After completion addition, reaction mixture was stirred for 1 h at room temperature and para-formaldehyde (40 g) was added. The resulting mixture was stirred further for 3 h at room temperature and then quenched with saturated aqueous NH4Cl solution. The organic layer was separated and aqueous layer was extracted with ethyl acetate (2 x 100 mL). The combined organic layers were washed with water, brine solution and dried over anhydrous Na2SO4. Concentration under reduced pressure and purification by silica gel column chromatography (EtOAc/pet.ether, 2:8) afforded alcohol 12 in 42.3 g, 85% yield as a viscous liquid. 1H NMR (CDCl3, 300 MHz): δ 7.21 (d, 2H, J = 8.3 Hz), 6.82 (d, 2H, J = 9.0 Hz), 4.44 (s, 2H), 4.17 (t, 2H, J = 2.2 Hz), 3.79 (s, 3H), 3.51 (t, 2H, J = 6.7 Hz), 2.51-2.44 (m, 2H).IR (Neat):3415, 2932, 2866, 1616, 1513, 1248, 1020, 1094, 822 cm-1. ESIMS: m/z 243 [M + Na]+.
To a stirred suspension of LiAlH4 (10.88 g, 286.36 mmol) in dry THF (40 mL) at 0°C was added dropwise a solution of 12 (42.0 g, 190.91 mmol) in dry THF (150 mL) under nitrogen. The reaction mixture was allowed to warm to room temperature and then refluxed for 4 h. It was then cooled to 0°C, diluted with ether and quenched by dropwise addition of saturated aqueous Na2SO4 (30 ml). The solid material was filtered and washed thoroughly with hot ethyl acetate several times. The combined organic layers were dried over anhydrous Na2SO4. Solvent was removed in vacuo and the residue was purified by silica gel column chromatography (EtOAc/pet.ether, 2:8) to afford allyl alcohol 13 (33.9 g, 80% yield) as a clear liquid. 1H NMR (CDCl3, 400 MHz): δ 7.12 (d, 2H, J = 8.9 Hz), 6.82 (d, 2H, J = 8.9 Hz), 5.67-5.61 (m, 2H), 4.39 (s, 2H), 4.02-3.98 (m, 2H), 3.78 (s, 3H), 3.44 (t, 2H, J = 6.4 Hz), 2.37-2.24 (m, 2H), 1.98 (brs, OH). IR (Neat):3449, 2935, 2835, 2855, 1736, 1609, 1512, 1246, 1032, 971 cm-1. ESIMS: m/z 245 [M + Na]+.
100ml dry DCM was added to 4°A powdered, activated molecular sieves (2 g) and the suspension mixture was cooled to –20°C. D (-) DET (6.12 g, 29.72 mmol) and Ti(OiPr)4 (9.38 mL, 29.72 mmol) were added subsequently with stirring and the resulting mixture was stirred for 30 min at –20°C. Allyl alcohol 13 (33.0 g, 148.64 mmol) in dry DCM (100 mL) was added and the resulting mixture was stirred for another 30 minutes at –20°C, cumenehydroperoxide (32 mL, 215.78 mmol) was added and the resulting mixture was stirred at the same temperature for 6 h. After completion of the reaction, (monitored by TLC) it was warmed to 0°C, quenched with 10 mL water and stirred for 1 h at 0°C. 30% aqueous NaOH solution saturated with NaCl (10 mL) was then added and the resulting mixture was stirred vigorously for another 30 min at 0°C. The resulting mixture was vacuum filtered through Celite and the filter cake was washed well with DCM. The organic phase was separated and aqueous phase was extracted with DCM (2 x 100 mL), the combined organic phases were washed with brine, dried over anhydrous Na2SO4. Removal of solvent under reduced pressure and purified by silica gel (EtOAc/pet.ether, 4:6) chromatography gave the epoxide 14 (24.7 g, 70 % yield), as a viscous liquid.[α]D25 : + 24.1 (c = 1, CHCl3). 1H NMR (CDCl3, 300 MHz): δ 7.19 (d, 2H, J = 8.5 Hz), 6.82 (d, 2H, J = 8.5 Hz), 4.41 (s, 2H), 3.78 (s, 3H), 3.62-3.47 (m, 4H), 3.07-2.98 (m, 1H), 2.98- 2.86 (m, 1H), 1.92-1.72 (m, 2H). 13C NMR (CDCl3, 75 MHz):δ 159.1, 130.1, 129.1, 113.7, 72.6, 66.4, 61.6, 56.4, 55.1, 53.6, 31.9. IR (Neat):3424, 2926, 2863, 1611, 1513, 1247, 1175, 1093, 1031, 819 cm-1. EIMS:m/z 239 [M + H]+.
To a stirred solution of 14 (24.0 g, 100.84 mmol) in a mixture of 90 mL of anhydrous ether and 10 mL of anhydrous CH3CN was added TPP (39.63 g, 151.12 mmol), imidazole (20.57 g, 302.52 mmol) and iodine (36.0 g, 121.10 mmol) at 0°C. The resulting mixture was stirred at room temperature for 20 min. After completion of reaction quenching with 10% aqueous Na2S2O3 solution and extracted with ether dried over anhydrous Na2SO4. The residue was concentrated under reduced pressure and purified by silica gel column chromatography (EtOAc/pet.ether, 1:9) to afford 15 as a colorless liquid (28.3 g, 81%).[α] D25 :– 16.2 (c = 1, CHCl3).1H NMR (CDCl3, 200 MHz): δ 7.21 (d, 2H, J = 8.7 Hz), 6.20 (d, 2H, J = 8.7 Hz), 4.42 (d, 2H, J = 3.9 Hz), 3.79 (s, 3H), 3.55-3.51 (m, 2H), 3.25-3.19 (m, 1H), 3.01-2.96 (m, 1H), 2.92-2.89 (m, 2H), 1.88-1.74 (m, 2H). IR (Neat):2930, 2859, 1611, 1512, 1247, 1174, 1096, 1033, 819 cm-1. ESIMS: m/z 349 [M +H]+.
A stirred suspension of 15 (28.0 g, 80.45 mmol) and zinc (52.2 g, 804.50 mmol) in anhydrous EtOH (100 mL) was refluxed for 30 min. The reaction mixture was filtered on Celite pad and concentrated under reduced pressure, crude product was purified by column chromatography (EtOAc/pet.ether, 3:7) to furnish 4(15.18 g, 85%)[α] D25 :– 9.3 (c = 1, CHCl3).1H NMR (CDCl3, 400 MHz): δ 7.19 (d, 2H, J = 7.8 Hz), 6.82 (d, 2H, J = 8.7 Hz), 5.83- 5.75 (m, 1H), 5.21 (d, 1H, J = 17.3 Hz), 5.07 (d, 1H, J = 10.5 Hz), 4.45 (s, 2H), 3.79 (s, 3H), 3.42 (dd, 2H, J = 2.9, 8.7 Hz), 3.29 (dd, 1H, J = 6.8, 8.7 Hz), 2.25 (br s, 1H), 2.22 (t, 2H, J = 6.8 Hz).13C NMR (CDCl3, 75 MHz): δ 155.1, 140.4, 129.9, 129.2, 114.2, 113.7, 72.8, 71.7, 67.9, 55.1, 36.2. IR (Neat):3443, 2934, 2861, 1612, 1512, 1246, 1092, 819 cm-1. ESIMS: m/z 245 [M + Na]+.
To a stirred solution of compound 4 (14.0 g, 63.06 mmol) in anhydrous dichloromethane (50 mL) at 0°C under nitrogen, iPr2NEt (32.9 mL, 189.18 mmol) was added followed by drop wise addition of MOMCl (9.5 mL, 126.12 mmol). After stirring for 2 h at room temperature, the reaction mixture was diluted with water, saturated aqueous NH4Cl, brine solution and then dried over anhydrous Na2SO4. The residue was concentrated under vacuo and purified by silica gel column chromatography (EtOAc/ pet.ether, 1:9) to afford the pure compound 16 (14.75 g, 88%) as a clear colorless liquid.[α]D25 :+ 44.8 (c = 1, CHCl3), 1H NMR (CDCl3, 500 MHz): δ7.20 (d, 2H, J = 8.3 Hz), 6.81 (d, 2H, J = 8.3 Hz), 5.69-5.62 (m, 1H), 5.16 (dd, 2H, J = 10.4, 18.7 Hz), 4.63 (d, 1H, J = 6.2 Hz), 4.47 (d, 1H, J = 6.2 Hz), 4.39 (s, 2H), 3.78 (s, 3H), 3.78-3.77 (m, 1H), 3.55-3.51 (m, 1H), 3.48-3.43 (m, 1H), 3.31 (s, 3H), 1.88-1.81 (m 1H), 1.79-1.72 (m, 1H). IR (Neat):2946, 1612, 1513, 1463, 1247, 1095, 1034, 994, 923, 820 cm-1. LCMS: m/z 267 [M + H]+.
To a stirred solution of compound 16 (14.0 g, 61.94 mmol) in DCM (27 mL) and water (3 mL) was added DDQ (16.8 g, 74.33 mmol) at room temperature. The reaction mixture was stirred for 2.5 h at room temperature before being quenched by the addition of 10 mL of saturated aqueous NaHCO3. The layers were separated and aqueous layer was extracted twice with DCM. The combined organic extracts were dried over anhydrous Na2SO4 and concentrated in vacuo. The crude product was purified by column chromatography on silica gel (EtOAc/pet.ether, 2:8) to give the compound 17 (5.7 g, 75%) as a pure yellow oil.[α] D25 :+ 54.2 (c = 1, CHCl3), 1H NMR (CDCl3, 300 MHz): δ5.76 (m, 1H), 5.23 (dd, 2H, J = 9.1, 14.3 Hz), 4.76 (d, 1H, J = 6.1 Hz), 4.51 (d, 1H, J = 6.7 Hz), 4.28-4.19 (m, 1H), 3.83-3.67 (m, 2H), 3.38 (s, 3H), 2.02 (br s,1H), 1.83-1.73 (m, 2H). IR(Neat): 3422, 2946, 2889, 1644, 1603, 1422, 1151, 1031, 925 cm-1. ESIMS: m/z 169 [M + Na]+.
To an ice-cold solution of iodoxybenzoic acid (15.8 g, 56.50 mmol) in DMSO (20 mL) was added a solution of alcohol 17 (5.5 g, 37.67 mmol) in dry DCM (20 mL). After stirring for 2 h at room temperature, the reaction mixture was filtered through a Celite pad and washed with ether. The combined organic layers were washed with water, brine solution and dried over anhydrous Na2SO4 and concentrated in vacuo. The crude product was purified by column chromatography on silica gel (EtOAc/pet.ether, 1:9) to give an aldehyde 18(3.9 g, 72%) as a viscous liquid. [α] D25 :+ 12.4 (c = 1, CHCl3),1H NMR (CDCl3, 200 MHz): δ 9.76 (t, 1H, J = 2.9 Hz), 5.77-5.71 (m, 1H), 5.28 (dd, 2H, J = 10.7, 29.2 Hz), 4.59 (ABq, 2H, J = 6.8 Hz), 4.58-4.53 (m, 1H), 3.33 (s, 3H), 2.71-2.05 (m, 1H), 2.55-2.49 (m, 1H).13C NMR (CDCl3, 75 MHz): δ200.4, 136.3, 118.0, 93.7, 72.0, 72.0, 55.4, 48.7. IR(Neat): 2927, 1721, 1638, 1421, 1217, 1030 cm-1.
A solution of the phosphonate S-I (9.3 g, 26.73 mmol) in dry THF (15 mL) was added to ice cold suspension of NaH (1.16 g, 48.61 mmol) in THF (10 mL). After the mixture was stirred for 30 min at 0°C, the reaction mixture was cooled to –78°C, and then a solution of aldehyde 18 (3.5 g, 24.30 mmol) in dry THF (15 mL) was added drop wise. After stirring for 1 h, reaction mixture was diluted with 10 mL of Et2O and quenched by the slow addition of 10 mL of H2O. The layers were separated, and the aqueous phase was extracted with two 10mL portions of Et2O. The organic extract was washed with brine solution, dried over anhydrous Na2SO4 and concentrated in vacuo. The crude product was purified by column chromatography on silica gel (EtOAc/ pet.ether, 0.5:9.5) to give an α, β-unsaturated ester 19 (4.1 g, 85%) as a viscous liquid. [α] D25: + 56.1 (c = 0.5, CHCl3)1H NMR (CDCl3, 200 MHz): δ6.31-6.26 (m, 1H), 5.81 (d, 1H, J = 10.7 Hz), 5.74-5.67 (m, 1H), 5.21 (dd, 2H, J = 9.8, 18.6 Hz), 4.65 (d, 1H, J = 6.8 Hz), 4.51 (d, 1H, J = 6.8 Hz), 4.14 (q, 2H, J= 6.8 Hz), 4.14- 1.10 (m, 1H), 3.34 (s, 3H), 2.93 (t, 3H, J = 6.8 Hz), 1.29 (t, 3H, J = 6.8 Hz). 13C NMR (CDCl3, 75 MHz): δ166.2, 145.3, 137.4, 121.2, 117.4, 93.8, 76.3, 59.8, 55.4, 34.6, 14.2. IR (Neat): 1612, 1720, 1384, 1248, 1033, 817 cm-1. ESIMS: m/z 237 [M + Na]+.
To a stirred solution of compound 19 (4 g, 32.25 mmol) in dry benzene (30 mL) was added catalytic amount of PTSA under nitrogen atmosphere. The reaction mixture was refluxed overnight. The aqueous layer was extracted twice with EtOAc, dried over anhydrous Na2SO4, and concentrated in vacuo. The crude product was purified by column chromatography on silica gel (EtOAc/pet.ether, 5:5) to give the lactone 2 (1.9 g, 85%) as a liqiud.[α] D25: (+)82.4 (c = 0.5, CHCl3); 1H NMR (CDCl3, 300 MHz): δ 6.83 (m, 1H), 6.02 (dd, 1H, J = 2.5, 11.1 Hz), 5.93 (m, 1H), 5.40 (d, 1H, J = 17.1 Hz), 5.28 (d, 1H, J = 11.1 Hz), 4.95-4.85 (m, 1H), 2.49-2.39 (m, 2H); 13C NMR (CDCl3, 75 MHz): δ 163.8, 144.4, 134.7, 121.4, 117.8, 77.7, 29.2; IR (neat): 2922, 1720, 1638, 1384, 1249, 1032, 763 cm-1; EIMS: 147 (M+ + 23).
In a 100 mL round bottomed flask, fitted with a nitrogen adaptor, the methyl (2S)-3-hydroxy-2-methylpropanoate 8 (5 g, 42.37 mmol) in dry CH2Cl2 (30 mL) was taken and catalytic amount of PTSA was added. Then the reaction mixture was cooled to 0°C. To this PMB imidate (14.28 g, 50.84 mmol) in dry CH2Cl2 was added drop wise. After completion of addition, the reaction mixture was allowed to stir for 3 h. The reaction mixture was diluted with CH2Cl2, and the organic layer was washed with water, aq. NaHCO3, dried over anhydrous Na2SO4. Concentration under reduced pressure and purification over silica gel column chromatography (EtOAc/hexane, 0.5:9.5) afforded pure 20 (9 g, 90%) as a viscous liquid.; [α] D25: (+) 4.1 (c = 1, CHCl3); 1H NMR (CDCl3, 300 MHz): δ 7.18 (d 2H, J = 8.6 Hz), 6.81 (d, 2H, J = 8.6 Hz), 4.41 (s, 2H), 3.79 (s, 3H), 3.67 (s, 3H), 3.57 (dd, 1H, J = 7.1, 8.8 Hz), 3.41 (dd, 1H, J = 6.1, 9.1 Hz), 2.77-2.65 (m, 1H), 1.15 (d, 3H, J = 7.7 Hz); IR (neat): 2945, 2850, 1736, 1611, 1512, 1247, 1178, 1089, 822 cm-1; EIMS; 256 (M+ + NH4+).
In two neck round bottomed flask weigh LiCl (4.65 g, 110.9 mmol) keep under nitrogen atmosphere, weigh NaBH4 (4.21 g, 110.9 mmol) and graind it to make powder very quikly and add to the above R.B. keeping R.B. in ice, add EtOH to the above mixture while stirring. Dissolve the compound 20 in freshly prepared dry THF and add slowly and gradually to the above reaction. Mixture being maintained at -10°C (ice+salt) check the TLC after 2 h. After completion of the reaction distill out EtOH and quench with saturated solution of NH4Cl and the organic layer was washed with water, aq. NaHCO3, dried over anhydrous Na2SO4. Concentration under reduced pressure and purification over silica gel column chromatography (EtOAc/hexane, 2:8) afforded pure 21 (6.9 g, 90%) as a viscous liquid; [α] D25: (+) 15.7 (c = 1, CHCl3); 1H NMR (CDCl3, 300 MHz): δ 7.19 (d, 2H, J = 8.6 Hz), 6.82 (d, 2H, J = 8.4 Hz), 4.42 (s, 2H), 3.79 (s, 3H), 3.61-3.51 (m, 2H), 3.48 (t, 1H, J = 4.5 Hz) 3.34 (t, 1H, J = 8.8 Hz), 2.42 (br, s, 1H), 2.01 (m, 1H), 0.87 (d, 3H, J = 6.9 Hz); IR (neat): 3415, 2867, 1612, 1512, 1247, 1035, 819 cm-1; EIMS: 233 (M+ + 23).
To an ice-cooled solution of iodoxybenzoic acid (13.59 g, 48.57 mmol) in DMSO (10 ml) was added a solution of alcohol 21 (6.8 g, 32.38 mmol) in dry CH2Cl2 (15 mL). After stirring for 2h at room temperature, the reaction mixture was filtered through a celite pad and washed with ether. The combined organic layers were washed with water, brine solution and dried over anhydrous Na2SO4 and concentrated in vacuo. The crude product was purified by column chromatography on silica gel (EtOAc/hexane, 1:9) to give an aldehyde 22(5.7 g, 85%) as a viscous liquid; [α] D25: (-)17.0 (c = 1, CHCl3); 1H NMR (CDCl3, 300 MHz): δ 9.68 (d 1H, J = 1.5 Hz), 7.18 (d, 2H, J = 9.1 Hz), 6.82 (d, 2H, J = 8.3 Hz), 4.42 (s, 2H), 3.79 (s, 3H), 3.62-3.54 (m, 2H), 2.59 (m, 1H), 1.11 (d, 3H, J = 7.5 Hz); IR (neat): 2960, 2855, 1709, 1611, 1513, 1248, 1091 cm-1; EIMS: 231 (M++ 23).
To a solution of (Methyl) triphenylphosphonium iodide (22.2 g, 54.63 mmol) in dry THF (30 mL), n-BuLi (20.3 mL, 32.62 mmol, 1.6M sol in n-Hexane) was added at 0°C under nitrogen atmosphere and stirred for 30 min. After that period, a clear red colored solution was obtained. At –78°C, a solution of aldehyde 22 (5.6 g, 27.18 mmol) in dry THF (20 mL) was added drop wise to the reaction mixture was allowed to stir at room temperature for 5 h. A saturated aqueous NH4Cl solution was added and extracted with EtOAc. The organic layer was washed with water, brine and dried over anhydrous Na2SO4. Solvent was removed under reduced pressure and purification by silica gel (EtOAc/ hexane, 0.4:9.6) column chromatography afforded 23 (3.32 g, 60%) as a viscous liquid; [α] D25 : (-)22.2 (c = 1, CHCl3); 1H NMR (CDCl3, 300 MHz): δ 7.19 (d, 2H, J = 8.3 Hz), 6.81 (d, 2H, J = 8.3 Hz), 5.81-5.69 (m, 1H), 5.01-4.96 (m, 2H), 4.41 (s, 2H), 3.79 (s, 3H), 3.34-3.19 (m, 2H), 2.51-2.41 (m, 1H), 1.02 (d, 3H, J = 6.7 Hz); 13CNMR (CDCl3, 75 MHz): δ 159, 141.2, 130.5, 129.1, 113.9, 113.6, 74.6, 72.5, 55.1, 37.7, 16.5.; IR (neat): 2959, 2854, 1612, 1515, 1247, 1092, 1036, 914 cm-1; EIMS: 229 (M+ + 23).
To a stirred solution of compound 23 (3.2 g, 15.53 mmol) in dry THF (15 mL) under N2 atmosphere was added BH3. Me2S (11.6 mL, 23.50 mmol) at 0°C. The mixture was allowed to warm to room temperature and stirred for 3 h. It was then cooled to 0°C and excess borane was quenched by careful addition of water. The reaction mixture was then treated with 20% aqueous NaOH solution (10 mL), 30% H202 and the resulting mixture was stirred at room temperature for 4 h. Excess H2O2 was quenched with saturated aqueous sodium metabisulphite solution and compound was extracted with EtOAc. The organic layer was washed with water, brine and dried over anhydrous Na2SO4. Removal of solvent under reduced pressure and purification by silica gel (EtOAc/hexane, 2:8) column chromatography afforded 24 (2.26 g, 65%) as a viscous liquid.; [α] D25: (+) 3.1 (c = 1, CHCl3); 1H NMR (CDCl3, 200 MHz): δ 7.20 (d, 2H, J = 8.7 Hz), 6.82 (d, 2H, J = 8.7 Hz), 4.42 (s, 2H), 3.79 (s, 3H), 3.67-3.54 (m, 2H), 3.34-3.21 (m, 2H), 2.52 (br, s, 1H), 1.92-1.86 (m, 1H), 1.61-1.51 (m, 2H), 0.93 (d, 3H, J = 7.3 Hz); IR (neat): 3415, 2867, 1612, 1512, 1247, 1035, 819 cm-1; EIMS: 225 (M+ + 1).
To a mixture of the alcohol 24 (2.1 g, 9.37 mmol) and imidazole (0.701 g, 10.31 mmol) in dry CH2Cl2 (10 mL) was added TBDPSCl (2.56 g, 9.37 mmoL) at 0°C. The mixture was stirred for 2h. at room temperature. The reaction mixture was diluted with water, washed with saturated aq NaCl and dried over anhydrous Na2SO4. The solvent was removed under vacuum and the residue was purified by column chromatography on silica gel (EtOAc/hexane, 1:9) to afford the pure silyl ether 25 (3.98 g, 92%) as a viscous liquid.; [α] D25: (-)17.0 (c = 1, CHCl3); 1H NMR (CDCl3, 200MHz): δ 7.65-7.59 (m, 4H), 7.39-7.29 (m, 6H), 7.16 (d, 2H, J = 8.3 Hz), 6.79 (d, 2H, J = 8.3 Hz), 4.35 (s, 2H), 3.78 (s, 3H), 3.67 (t, 2H, J = 6.7 Hz), 3.27-3.14 (m, 2H), 1.99-1.89 (m, 1H), 1.75-1.64 (m, 1H), 1.41-1.31 (m, 1H), 1.03 (s, 9H), 0.89 (d, 3H, J = 6.7 Hz); 13 CNMR (CDCl3, 75MHz): δ 135.5, 134.7, 129.6, 129.4, 129, 127.6, 127.5, 113.6, 75.5, 72.4, 62, 55.2, 36.4, 30.2, 26.8, 26.5, 19.1, 17.2; IR (neat): 2936, 2858, 1612, 1466, 1246, 1105, 819 cm-1; EIMS: 480 (M+ + NH4+).
To a stirred solution of compound 25 (3.8 g, 8.22mmol) in CH2Cl2 (15 mL) and water (2 mL) was added DDQ (2. 24 g, 9.87 mmol) at room temperature. The reaction mixture was stirred for 2.5 h at room temperature before being quenched by the addition of 10 mL of saturated aqueous NaHCO3. The layers were separated and aqueous layer was extracted twice with CH2Cl2. The combined organic extracts were dried over anhydrous Na2SO4 and concentrated in vacuo. The crude product was purified by column chromatography on silica gel (EtOAc/hexane, 1:9) to give the compound 26 (2.39 g, 85%) as a pure yellow oil.; [α] D25: (+) 6.8 (c = 1, CHCl3); 1H NMR (CDCl3, 400 MHz): δ 7.66 (m, 4H), 7.41- 7.35 (m, 6H), 3.78-3.64 (m, 2H), 3.51-3.41 (m, 2H), 2.29 (br,s, 1H), 1.88-1.77 (m, 1H), 1.67-1.56 (m, 1H), 1.53-1.41 (m, 1H), 1.05 (s, 9H), 0.91 (d, 3H, J = 6.7 Hz); IR (neat): 3349, 3062, 2938, 2868, 1468, 1428, 1106, 1003, 815, 702 cm-1; EIMS: 343 (M+ + 1).
To an ice-cooled solution of iodoxybenzoic acid (2.71 g, 9.67 mmol) in DMSO (5 ml) was added a solution of alcohol 26(2.2 g, 64.3 mmol) in dry CH2Cl2 (10 mL). After stirring for 2 h at room temperature, the reaction mixture was filtered through a celite pad and washed with ether. The combined organic layers were washed with water, brine solution and dried over anhydrous Na2SO4 and concentrated in vacuo. The crude product was purified by column chromatography on silica gel (EtOAc/hexane, 0.5:9.5) to give an aldehyde 27(1.9 g, 82%) as a viscous liquid.; [α] D25: (-) 28.4 (c = 1, CHCl3); 1H NMR (CDCl3, 400 MHz): δ 9.64 (d, 1H, J = 1.5 Hz), 7.66-7.58 (m, 4H), 7.41-7.31 (m, 6H), 3.75-3.62 (m, 2H), 2.61-2.51 (m, 1H), 2.06-1.93 (m, 1H), 1.65-1.51 (m, 1H), 1.08 (d, 3H, J = 7.5 Hz), 1.03 (s, 9H).; 13C NMR (CDCl3, 75 MHz): d 104.5, 135.5, 134.7, 133.1, 129.7, 127.6, 65.5, 64, 48.7, 42, 26.6, 19.2, 13.2, 10.2; IR (neat): 2932, 2859, 1707, 1107, 702 cm-1; EIMS: 358 (M+ + NH4+).
A solution of the phosphonium salt S-II (1.94 g, 5.17 mmol) in dry THF (5 mL) was added over a cooled (0ºC) suspension of NaH (0.225 g, 9.41 mmol) in THF (5 mL). After the mixture was stirred for 30 min at 0°C, the reaction mixture was cooled to –78 °C, and then a solution of aldehyde 27 (1.6 g, 4.70 mmol) in dry THF (6 mL) was added drop wise. After stirring for 1 h and the reaction mixture was diluted with 5 mL of Et2O and quenched by the slow addition of 4mL of H2O. The layers were separated, and the aqueous phase was extracted with two 10 mL portions of Et2O, The organic extract was washed with brine solution, dried over anhydrous Na2SO4 and concentrated in vacuo. The crude product was purified by column chromatography on silica gel (EtOAc/hexane, 0.3:9.7) to gave α,β-unsaturated ester 28 (1.81 g, 88%) as a viscous liquid.; [α] D25: (+) 25.6 (c = 1, CHCl3); 1H NMR (CDCl3, 300 MHz): δ 7.66-7.61 (m, 4H), 7.41-7.33 (m, 6H), 5.51 (d, 1H, J = 10.1 Hz), 4.14 (q, 2H, J = 7.1, 14.3 Hz), 3.61 (dt, 2H, J = 1.3, 5.8 Hz), 3.18-3.07 (m, 1H), 2.21 (q, 2H, J = 7.5, 15.2 Hz), 1.62-1.51 (m, 2H), 1.24 (t, 3H, J = 7.1Hz), 1.02 (s, 9H), 0.99 (d, 3H, J = 3.3 Hz), 0.96 (t, 3H, J = 2.6 Hz); 13C NMR (CDCl3, 75 MHz): δ 168.2, 144.9, 135.5, 133.9, 132.7, 129.4, 127.5, 62.2, 59.9, 40, 30.2, 27.2, 26.7, 20.8, 20.4, 19.2, 14.2, 13.6; IR (neat): 2961, 2932, 1713, 1644, 1107, 703 cm-1; EIMS: 439 (M+ + 1).
At -78°C 1.4M solution of DIBAL-H (4.15 mL, 7.30 mmol) was slowly added to a solution of α,β-unsaturated ester 28 (1.6 g, 3.65 mmol) in dry CH2Cl2 (10 mL). The solution was stirred for 2 h at -78°C before being quenched with EtOAc (5 mL). The mixture was allowed to warm to ambient temperature before an aqueous solution of Rochelle’s salt was added (30 mL) and stirred for one hour. The aqueous phase was extracted with CH2Cl2 and the combined organic extracts were dried over anhydrous Na2SO4, concentrated, and purified by column chromatography on silica gel (EtOAc/hexane, 1.5:8.5) to afford the the desired allylic alcohol 29 (1.31 g, 90%) as a pale yellow oil.; [α] D25: (-)12.4 (c = 1, CHCl3); 1H NMR (CDCl3, 200 MHz): δ 7.65-7.59 (m, 4H), 7.41- 7.32 (m, 6H), 4.90 (d, 1H, J = 10.1 Hz), 4.22 (d, 1H, J = 11.7 Hz ), 3.89 (dd, 1H, J = 4.9, 12.8 Hz), 3,71-3.55 (m, 2H), 2.896-2.73 (m, 1H), 2.12 (q, 2H, J = 7.3, 14.9 Hz), 1.88-1.74 (m, 1H), 1.53- 1.45 (m, 1H), 1.05 (s, 9H), 1.02 (t, 3H, J = 3.2 Hz), 0.92 (d, 3H, J = 6.7 Hz); 13C NMR (CDCl3, 75 MHz): δ 140.1, 135.6, 135.5, 132.6, 129.7, 129.6,127.6, 62, 60.6, 39.9, 28.3, 28.1, 26.8, 21.8, 19.1, 12.9; IR(KBr): 3429, 3068, 2930, 2861, 1631, 1464, 1427, 1106, 1101,702 cm-1; EIMS: 397(M+ + 1).
To an ice-cooled solution of iodoxybenzoic acid (1.27 g, 4.54 mmol) in DMSO (5 ml) was added a solution of alcohol 29(1.2 g, 3.03 mmol) in dry CH2Cl2 (10 mL). After stirring for 2 h at room temperature, the reaction mixture was filtered through a celite pad and washed with ether. The combined organic layers were washed with water, brine solution and dried over anhydrous Na2SO4 and concentrated invacuo. The crude product was purified by column chromatography on silica gel (EtOAc/hexane, 1:9) to give an aldehyde 30 (0. 954 g, 80%) as a viscous liquid.; [α] D25 : (-) 6.9 (c = 1, CHCl3); 1H NMR (CDCl3, 300 MHz): δ 10.12 (s, 1H), 7.79 (dd, 4H, J = 6.8, 14.6 Hz), 7.41-7.31 (m, 6H), 6.08 (d, 1H, J = 10.7 Hz), 3.65-3.55 (m, 2H), 3.54-3.48 (m, 1H), 2.15 (q, 2H, J = 7.8, 15.6 Hz), 1.73-1.66 (m, 1H), 1.49-1.43 (m, 1H), 1.07 (d, 3H, J = 5.8 Hz), 1.03 (s, 9H), 0.96 (t, 3H, J = 7.8 Hz); 13C NMR (CDCl3, 75 MHz): δ 191.3, 153.5, 140.7, 135.4, 133.5, 129.6, 127.6, 61.3, 39.8, 27.3, 26.7, 21.3, 19, 13.1; IR (neat): 2960, 2831, 1726, 1146, 1106, 702 cm-1; EIMS: 12 (M+ + NH4+).
To a solution of (Methyl) triphenylphosphonium iodide (1.66 g, 4.06 mmol) in dry THF (10 mL), n-BuLi (1.5 mL, 2.43 mmol, 1.6M sol in n-Hexane) was added at 0°C under nitrogen atmosphere and stirred for 30 min. After that period, a clear red colored solution was obtained. At –78°C, a solution of aldehyde 30 (0.8 g, 2.03 mmol) in dry THF (5 mL) was added drop wise to the reaction mixture was allowed to stir at room temperature for 5h. A saturated aqueous NH4Cl solution was added and extracted with EtOAc. The organic layer was washed with water, brine and dried over anhydrous Na2SO4. Solvent was removed under reduced pressure and purification by silica gel (EtOAc/hexane, 0.3:9.7) column chromatography afforded 31 (0.516 g, 65%) as a viscous liquid. [α] D25: (-) 4.4 (c = 1, CHCl3); 1H NMR (CDCl3, 400MHz): δ 7.64-7.59 (m, 4H), 7.38-7.29 (m, 6H), 6.69 (dd, 1H, J = 11.3, 17.4 Hz), 5.19 (d, 1H, J =17.4 Hz), 5.02 (dd, 2H, J = 9.2, 21.5 Hz), 3.59 (t, 2H, J = 7.1 Hz), 2.92-2.84 (m, 1H), 2.16 (q, 2H, J = 7.1, 14.3 Hz), 1.63-1.56 (m, 1H), 1.46-1.36 (m, 1H), 1.03 (s, 9H), 1.02 (t, 3H, J = 7.1 Hz), 0.95 (d, 3H, J = 6.1 Hz).; 13C NMR (CDCl3, 75 MHz): δ 137, 135.5, 134, 133.2, 129.4, 127.5, 112.6, 61.8, 40.7, 27.8, 26.9, 25.8, 21.3, 19.1, 13.4.; IR (neat): 2959, 2930, 2861, 1640, 1592, 1463, 1107, 988, 702 cm-1. EIMS: 393 (M+ + 1).
To a solution of the compound 31 (0.4 g, 1.02 mmol) in THF (5 mL) was added 1M solution of n-Bu4NF (1.22 mL) at 0 °C. The reaction mixture was stirred at room temperature for 1 h. After completion of the reaction diluted with ether. The combined organic layers were washed with saturated aqueous NaCl, dried over anhydrous Na2SO4 and concentrated under reduced pressure. The resulted crude product was purified on silica gel column chromatography (EtOAc/hexane, 1:9) to afford a colorless liquid 5 (0.133g, 85%).; [α] D25: (-)30.0 (c = 1, CHCl3); 1H NMR (CDCl3, 300 MHz): δ 6.66 (dd, 1H, J = 10.7, 17.5 Hz), 5.22 (d, 1H, J = 17.5 Hz), 5.09 (dd, 2H, J = 10.7, 20.5 Hz), 3.62-3.52 (m, 2H), 2.83-2.76 (m, 1H), 2.20 (q, 2H, J = 7.8, 15.6 Hz), 1.66-1.59 (m, 1H), 1.48- 1.4 (m, 1H), 1.06 (t, 3H, J = 7.8 Hz), 0.99 (d, 3H, J = 6.8 Hz); 13C NMR (CDCl3, 75 MHz): δ 139.8, 134.9, 132.7, 113.8, 61.2, 40.3, 28.3, 25.8, 21.5, 13.4; IR (neat): 3484, 2962, 2873, 1639, 1457, 1057, 993, 902 cm-1; EIMS: 177 (M+ + 23).
A 250 ml flask equipped with a septum inlet, a magnetic stirring bar, was charged with 5.05 mL of BH3. SMe2 (50 mmol) and 18 mL of THF. It was cooled to 0°C and 18.3 mL (115 mmol) of (+)-α-pinene (neat) was added drop wise. After the mixture was stirred at 0°C for 1 h. [(-)-Ipc2BH separated as a white solid during this time] the flask was stored in a refrigerator at 0°C for 2 days. To the (-)-Ipc2BH (solid, 50 mmol) was added neat olefin 32 (18.3 g, 75 mmol). The reaction mixture was stirred at –25°C for 1 h. and ept in the refrigerator for 5 days. The trialkyl borane was treated with 50 mL of 3N sodium hydroxide, 7.5 mL of 30% hydrogen peroxide and stirred at 25°C for 5 h. Compound was extracted with ether, dried over Na2SO4 and the ether was evaporated. The residue was filtered through silica gel (pet.ether-ethyl acetate, 9:1 used as eluent) to remove the olefin and α-pinene alcohol and then eluted with pet.ether-ethyl acetate (1:1) mixture to give the pure alcohol 33 in 90% yield; [α] D25: (-)3.6 (c = 6.5, CHCl3); 1H NMR (CDCl3, 200 MHz): δ 7.41-7.20 (m, 5H), 4.72-4.60 (m, 1H), 4.48 (s, 2H), 4.12 (dd, 1H, J = 3.8, 10.5 Hz), 3.80 (d, 1H, J = 3.8 Hz), 3.40 (t, 1H, J = 3.2 Hz), 2.85 (dd, 1H, J = 6.6, 12.5 Hz), 2.35 (brs, 1H, OH), 1.60–1.45 (m, 1H), 2.10–1.95 (m, 2H), 1.07 (d, 3H, J = 6.7 Hz), 0.92 (d, 3H, J = 6.8 Hz); IR (Neat): 3440 cm-1; FAB mass: m/z 262 (M+).
Pyridinium chlorochromate (PCC) (16.12 g, 75 mmol) was added to a solution of alcohol 33 (13.1g, 50 mmol) in CH2Cl2 (150 mL). After stirring the reaction mixture for 3 h. isopropanol (10 mL) was added and the solvent was removed under reduced pressure. The residue was triturated with ether and the organic layer was washed with dil. HCl, water, brine solution and dried over Na2SO4. Removal of solvent afforded a gummy material which was crystallized from pet ether-benzene (9:1) mixture to afford the ketone 34 (11.05 g, 85%) as a solid. [α] D25: (+) 23.9 (c = 4.5, CHCl3).;1H NMR (CDCl3, 200 MHz): δ 7.50–7.25 (m, 5H), 4.55 (ABq, 2H, J = 10.3 Hz), 4.31 (dd, 1H, J = 3.7, 8.8 Hz), 3.65–3.50 (m, 2H), 2.85–2.75 (m, 1H), 2.20-2.40 (m, 3H), 1.08 (d, 3H, J = 6.8 Hz), 0.95 (d, 3H, J = 6.8 Hz); IR (Neat): 1760 cm-1; EI mass: m/z 260 (M+); m.p : 61°C.
To a suspension of NaHCO3 (10.6 g, 64.0 mmol) in CH2Cl2 (60 mL) was added the ketone 34 (11 g, 43.0 mmol) in CH2Cl2 (30 mL) under nitrogen atmosphere. Then, dry m-CPBA (11.14 g, 64.0 mmol) was added in small fractions to the reaction mixture at 0°C. It was stirred at ambient temperature for 10 h. TLC monitored completion of the reaction. The reaction mixture was diluted with dichloromethane and the CH2Cl2 layer was washed with a solution of sodium metabisulphite followed by 5% NaHCO3 solution and water. The organic layer was dried over anhydrous Na2SO4 and concentrated to dryness under reduced pressure. The residue was purified by silica gel chromatography to afford the pure lactone 35 (10.68 g, 90%) as a oil.; [α] D25: (-) 46.2 (c = 6.5, CHCl3); 1H NMR (CDCl3, 200 MHz): δ 7.70–7.35 (m, 5H), 5.46 (s, 1H), 4.60 (ABq, 2H, J = 10.6 Hz), 4.11 (m, 1H), 3.61 (t, 1H, J = 5 Hz), 2.75–2.65 (m, 2H), 2.25–2.15 (m, 1H), 2.09–1.98 (m, 1H), 1.18 (d, 3H, J = 7.6 Hz), 0.98 (d, 3H, J = 7.6 Hz); IR (Neat): 1755 cm-1; FAB mass: m/z 276 (M+).
LDA was prepared by the addition of n-BuLi (1.6 molar solutionin hexane, 27.1 mL, 43.4 mmol) to a 0°C cooled solution of DIPA (6.62 mL, 47 mmol)] in dry THF (18 mL). After stirring at 0°C for 45 min, it was added to a solution of lactone 35 (10 g, 36.2 mmol) in dry THF (40 mL) at –78°C. After 1 h, the lithium enolate thus generated was alkylated with ethyl iodide (8.7 mL, 108.6 mmol). Stirring was continued for further 2 h at –78°C and 2h at room temperature and quenched with saturated ammonium chloride. The reaction mixture was extracted with ether, dried over anhydrous Na2SO4, evaporated the solvent and on purification by column chromatography on silica gel gave the ethylated lactone 36 (9.29 g, 85%) as a viscous liquid; [α] D25: (-) 49.0 (c = 1, CHCl3).; 1H NMR (CDCl3, 300 MHz): δ 7.33–7.23 (m, 5H), 5.34 (d, 1H, J = 3.0 Hz), 4.64 (d, 1H, J = 12.0 Hz), 4.48 (d, 1H, J = 12.0 Hz), 3.76 (d, 1H, J = 4.5 Hz), 3.55 (t, 1H, J = 3.0 Hz), 2.51 (t, 1H, J = 7.5 Hz), 2.19–2.14 (m, 1H), 1.99–2.03 (m, 1H), 1.81 (quintet, 2H, J = 7.5 Hz), 1.13 (d, 3H, J = 7.5 Hz), 0.99 (t, 3H, J = 7.5 Hz), 0.91 (d, 3H, J = 7.5 Hz); 13C NMR (CDCl3, 75 MHz): δ 169.7, 137.7, 129.8, 128.0, 127.3, 99.7, 79.1, 76.5, 73.8, 42.1, 39.6, 37.4, 26.8, 13.3, 13.0, 11.5; IR (neat): 2969, 1730, 1212, 771 cm-1; FAB Mass: m/z 305 (M++1).
To a stirred suspension of LiAlH4 (1.31 g, 34.5 mmol) in dry THF (30 mL) at 0°C, a solution of lactone 36 (6.9 g, 23.0 mmol) in dry THF (30 mL) was added drop wise. The reaction mixture was refluxed for 4 h. It was then cooled to 0°C, diluted with ether and quenched with drop wise addition of saturated aqueous Na2SO4. The solid material was filtered and washed thoroughly with hot ethyl acetate for several times. The combined organic layers were dried over anhydrous Na2SO4. The solvent was removed under vacuo and the residue was purified by silica gel column chromatography to afford the compound 9 (5.7 g, 80%) as a viscous liquid. [α]D25: (-) 4.5 (c = 1, CHCl3); 1H NMR (CDCl3, 300 MHz): δ 7.30 (m, 5H), 4.65 (s, 2H), 3.96–3.49 (m, 6H), 2.07–1.84 (m, 2H), 1.65–1.51 (m, 1H), 1.29–1.17 (m, 1H), 1.12 (d, 3H, J = 7.5 Hz), 1.10–0.98 (m, 1H), 0.96 (d, 3H, J = 7.5 Hz), 0.92 (t, 3H, J = 6.7 Hz); 13C NMR (CDCl3, 75 MHz): δ = 137.5, 128.6, 128.1, 127.8, 127.1, 88.5, 76.3, 74.9, 65.3, 65.0, 43.8, 37.8, 35.5, 20.7, 14.7, 11.7, 11.3; IR (neat): 3419, 3295, 2963, 1042 cm-1; FAB mass: m/z 311 (M++1).
To a solution of triol 9 (5.5 g, 17.7 mmol) in dry CH2Cl2 (40 mL), 2, 2-dimethoxy propane (16 mL, 123.9 mmol) and PTSA (2.2 g, 14.1 mmol) was added. The mixture was stirred at ambient temperature for 12 h. Sodium bicarbonate was added to neutralize PTSA and filtered. Removal of solvent and purification by silica gel column chromatography afforded the mono acetonide 37(4.9 g, 80%) as a white solid.; [α]D25: (-) 38.0 (c = 1, CHCl3); 1H NMR (CDCl3, 300 MHz): δ 7.24–7.34 (m, 5H), 4.64 (ABq, 2H, J = 11.3, 29.4 Hz), 3.43–3.96 (m, 6H), 2.60 (brs, OH), 1.86–2.06 (m, 2H), 1.64–1.75 (m, 1H), 1.36–1.44 (m, 1H), 1.33 (s, 6H), 1.21 (d, 3H, J = 7.5 Hz), 0.96–1.09 (m, 1H), 0.90 (d, 3H, J = 7.5 Hz), 0.86 (t, 3H, J = 6.7 Hz); IR (neat): 3478, 2921, 1617, 1031 cm-1; FAB mass: m/z 351 (M++1); m.p : 72°C
To a stirred and cooled solution of alcohol 37 (4.5 g, 12.8 mmol) and Pyridine (5.19 mL, 64.2 mmol) in Dry DCM (20 mL), trimethyl acetyl chloride (1.85 g, 15.3 mmol) was added slowly. It was stirred overnight at room temperature. The reaction mixture was diluted with DCM and washed with aqueous copper sulphate solution, water, and brine solution and dried over anhydrous Na2SO4. The solvent was removed under vacuo and the residue was purified by silica gel column chromatography afforded the compound 38 (4.72 g, 85%) as a liquid.; [α]D25: (-) 20.0 (c = 1, CHCl3); 1H NMR (CDCl3, 300 MHz): δ 7.34–7.24 (m, 5H), 4.64 (ABq, 2H, J = 11.7, 18.1 Hz), 4.32 (dd, 1H, J = 5.2, 10.9 Hz), 3.95 (d, 1H, J = 8.1 Hz), 3.91 (d, 1H, J = 8.1 Hz), 3.82 (dd, 1H, J = 5.2, 10.9 Hz), 3.51 (t, 1H, J = 10.9 Hz), 3.42 (dd, 1H, J = 1.8, 10.9 Hz), 2.24–2.16 (m, 1H), 2.01-1.94 (m, 1H), 1.75–1.68 (m, 1H), 1.36 (s, 3H), 1.33 (s, 3H), 1.24–1.20 (m, 2H), 1.22 (s, 9H), 1.15 (d, 3H, J = 6.9 Hz), 0.94 (d, 3H, J = 6.9 Hz), 0.89 (t, 3H, J = 7.3 Hz); 13C NMR (CDCl3, 75 MHz): δ 178.6, 139.1, 128.2, 127.1, 126.6, 97.7, 82.7, 74.8, 71.6, 65.7, 64.0, 38.7, 37.0, 36.4, 34.9, 29.6, 27.1, 26.9, 20.6, 19.7, 16.2, 10.7, 9.8; IR (neat): 2960, 1720, 1100 cm-1; FAB mass: m/z 435 (M++1).
To a stirred solution of compound 38 (4.5 g 10.3 mmol) in methanol (30 mL) was added catalytic amount of PTSA. The reaction mixture was stirred 10 h. at room temperature. Sodium bicarbonate was added to neutralize PTSA and filtered. The filtrate was concentrated under reduced pressure and purification by silica gel column chromatography afforded 39 (3.04 g, 75%) as a pure liquid. [α] D25: (-)5.5 (c = 1, CHCl3); 1H NMR (CDCl3, 300 MHz): δ 7.34-7.26 (m, 5H), 4.57 (AB q, 2H, J = 10.5, 21.1 Hz), 4.26 (dd, 1H, J = 3.0, 10.5 Hz), 4.14 (dd, 1H, J = 5.2, 10.5 Hz), 3.89 (d, 1H, J = 9.8 Hz), 3.76 (brs, -OH), 3.69-3.57 (m, 2H), 3.42 (dd, 1H, J = 2.2, 9.0 Hz), 3.23 (brs, -OH), 2.26–2.17 (m, 1H), 1.96- 1.88 (m, 1H), 1.64-1.54 (m, 1H), 1.11-1.01 (m, 2H), 1.23 (s, 9H), 1.15 (d, 3H, J = 6.8 Hz), 0.96 (d, 3H, J = 6.8 Hz), 0.93 (t, 3H, J = 6.8 Hz); IR (neat): 3452, 2969, 1726, 1160, 976 cm-1; FAB mass: m/z 395 (M++1).
A solution of 39 (2.8 g, 7.1 mmol) in dry CH2Cl2 (15 mL), containing triethyl amine (1.48 g, 10.6 mmol) was cooled to 0°C and treated with p-toluenesulphonyl chloride (2.02 g, 10.6 mmol) and catalytic amount of Bu2SnO. The reaction mixture was stirred at room temperature for 10 h. It was diluted with water and extracted with CH2Cl2. The organic layer was washed with brine and dried over anhydrous Na2SO4. Removal of solvent under reduced pressure and purification by silica gel column chromatography afforded 40 (2.72 g, 70%) as a viscous liquid; [α] D25: (+) 10.0, (c = 1, CHCl3); 1H NMR (CDCl3, 300 MHz): δ 7.75 (d, 2H, J = 8.4 Hz), 7.26–7.33 (m, 5H), 7.24 (d, 2H, J = 8.4 Hz), 4.53 (q, 2H, J = 10.1 Hz), 4.06–4.28 (m, 4H), 3.72 (d, 1H, J = 10.1 Hz), 3.39 (dd, 1H, J = 3.3, 8.4 Hz), 3.06 (d, 1H, J = 1.6 Hz), 2.39 (s, 3H), 2.06-2.19 (m, 1H), 1.80–1.93 (m, 1H), 1.48–1.63 (m, 1H), 1.18-1.27 (m, 2H), 1.23 (s, 9H), 1.02 (d, 3H, J = 6.7 Hz), 0.96 (d, 3H, J = 6.7 Hz), 0.87 (t, 3H, J = 7.6 Hz); IR (neat): 3498, 2972, 1726, 1359, 1174 cm-1; FAB mass: m/z 549 (M++1).
tert-butyldimethylsilyl trifluoromethanesulfonate (1.28 g, 5.47 mmoL) was added to an ice-cold solution of compound 40 (2.5 g, 4.56 mmol) in dry CH2Cl2 (15 mL) followed by the slow addition of 2,6-lutidine (1.59 g, 13.68 mmoL). The reaction mixture was stirred for 1 h at 0°C before being diluted with EtOAc and washed with saturated aqueous NH4Cl and brine solution. The organic phase was dried over Na2SO4, and concentrated. Purification by column chromatography afforded the pure compound 41 (2.56 g, 85%); [α]D25: (+) 5.4 (c = 0.6, CHCl3); 1H NMR (CDCl3, 200MHz): d 7.72 (d, 2H, J = 8.1 Hz), 7.33-7.29 (m, 7H), 4.57 (s, 2H), 4.27-4.07 (m, 2H), 3.98-3.89 (m, 3H), 3.33-3.27 (m, 1H), 2.44 (s, 3H), 2.16-1.68 (m, 3H), 1.34-1.41 (m, 2H), 1.20 (s, 9H), 1.10 (s, 3H), 0.88 (d, 3H, J = 6.8 Hz), 0.86 (s, 9H), 0.80 (t, 3H, J = 7.6 Hz), 0.06 (s, 3H), 0.01 (s, 3H); IR (neat): 2970, 1723, 1300, 1170 cm-1; FAB Mass: m/z 663 (M++1).
To a stirred suspension of LiAlH4 (0.3 g, 6.04 mmol) in dry THF (10 mL) at 0°C was added drop wise a solution of compound 41 (2.0 g, 3.02 mmol) in dry THF (15 mL). The reaction mixture was refluxed for 4 h. It was then cooled to 0°C, diluted with ether and quenched with drop wise addition of saturated aqueous Na2SO4. The solid material was filtered and washed thoroughly with hot ethyl acetate for several times. The combined organic layers were dried over anhydrous Na2SO4. The solvent was removed under vacuo and the residue was purified by silica gel column chromatography to afford the compound 42 (0.98 g, 80%) as a viscous liquid.; [α]D25: (+) 2.5 (c = 1, CHCl3); 1H NMR (CDCl3, 200MHz): δ 7.31–7.24 (m, 5H), 4.61 (brs, 2H), 3.75 (t, 2H, J = 2.9 Hz), 3.56–3.45 (m, 1H), 3.34–3.28 (dd, 1H, J = 3.7, 7.4 Hz), 2.60–2.51 (m, 1H), 2.01–1.85 (m, 2H), 1.56–1.40 (m, 2H), 1.14 (d, 3H, J = 6.6 Hz), 0.90 (s, 9H), 0.87 (d, 3H, J = 6.6 Hz), 0.85 (t, 3H, J = 7.4 Hz), 0.82 (d, 3H, J = 6.6 Hz), 0.05 (s, 3H), 0.03 (s, 3H); IR (neat): 3476, 3032, 2959, 1092, 1050 cm-1.; FAB mass: m/z 409 (M++1).
To a freshly distilled ammonia (20 mL) in 50 mL two neck round bottom flask fitted with a cold finger condenser, was added lithium metal (0.15 g, 22.0 mmol) in fractions at –33°C and the resulting gray colored suspension was stirred for 30 min. To this was added compound 42 (0.9 g, 2.2 mmol) in dry THF (10 mL) over a period of 10 min. The reaction mixture was then stirred for another 30 min at –33°C and quenched by the addition of solid ammonium chloride and the ammonia was then allowed to evaporate. The residue left was partitioned between water and ether and the aqueous phase extracted with ether. The organic layers were combined, washed once with water, brine, dried over anhydrous Na2SO4, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography to afford the pure 43 (0.56 g, 80%) as a clear colorless liquid.; [α]D25: (+)2.7 (c = 1.1, CHCl3); 1H NMR (CDCl3, 200 MHz): δ 4.25 (br, OH), 3.93 (dd, 1H, J = 11.0, 3.0 Hz), 3.83 (t, 1H, J = 2.7 Hz), 3.71 (dd, 1H, J = 9.1, 3.0 Hz), 3.57 (dd, 1H, J = 11.0, 4.3 Hz), 3.15 (br, OH), 2.08–2.00 (m, 1H), 1.76–1.68 (m, 1H), 1.62-1.55 (m, 1H), 1.47–1.37 (m, 1H), 1.26–1.16 (m, 1H) 1.14(d, 3H, J = 7.0 Hz), 0.97 (d, 3H, J = 6.7 Hz), 0.92 (s, 9H), 0.89 (t, 3H, J = 7.3 Hz), 0.83 (d, 3H, J = 7.0 Hz) 0.14 (s, 3H), 0.10 (s, 3H); 13C NMR (CDCl3, 75 MHz): δ 80.2, 79.7, 64.5, 40.1, 36.5, 35.6, 28.4, 25.8, 15.5, 15.2, 13.8, 12.2, -4.2, -4.5; IR (neat): 3413, 2959, 1024 cm-1; FAB mass: m/z 319 (M++1).
In an oven-dried flask under N2 atmosphere DMSO (0.28 mL, 3.93 mmol) was dissolved in dry CH2Cl2 (5 mL). The solution was cooled to –78°C, and (COCl)2 (0.25 g, 1.96 mmol) was added drop wise. After 5 min diol 43 (0.5 g, 1.57 mmol) in dry CH2Cl2 (5 mL) was added drop wise. The white slurry was stirred for 10 min at –78°C, before Et3N (0.79 g, 7.85 mmol) was added drop wise. The solution was allowed to warm to ambient temperature before being diluted in Et2O (10 mL) and washed with aqueous NH4Cl (10 mL), NaHCO3 (10 mL), and brine (10 mL). The organic phase was dried over anhydrous Na2SO4 and concentrated invacuo to give crude hydroxy aldehyde (0.39 g, 80%), which is directly used for next reaction.
Mixture of above prepared aldehyde (0.39 g, 1.23 mmol) and (1-ethoxy carbonylethylidene)triphenyl phosphorane salt (1.12 g, 3.08 mmol) in dry CH2Cl2 (5 mL) was refluxed and stirred for 15 h. The reaction mixture was concentrated and the resultant residue was purified by column chromatography to give α,β- unsaturated ester 44 (0.39 g, 80%) as a solid.; [α]D25(+) 30.9 (c = 1, CHCl3); 1H NMR (CDCl3, 400 MHz): δ 6.98 (dq, 1H, J = 10.5, 1.2 Hz), 4.2-4.35 (brs, 1H), 4.2-4.11 (q, 2H, J = 6.6 Hz), 3.7 (t, 1H, J = 3.5 Hz), 3.61 (d, 1H, J = 3.5 Hz), 2.64-2.54 (m, 1H), 1.80 (d, 3H, J = 1.4 Hz) 1.65-1.48 (m, 2H), 1.39-1.17 (m, 2H), 1.28 (t, 3H, J = 7.1 Hz), 1.11(d, 3H, J = 7.1 Hz), 0.95 (d, 3H, J = 6.6 Hz), 0.85 (s, 9H), 0.80 (t, 3H, J = 7.5 Hz), 0.71 (d, 3H, J = 7.0 Hz) 0.09 (s, 3H), 0.01 (s, 3H); 13C NMR (CDCl3, 75 MHz): δ 168.2, 142.5, 127.5, 80.7, 60.2, 41.6, 36.3, 35.6, 29.0, 25.8, 16.7, 15.3, 14.2, 13.8, 12.4, 12.0, -4.1, -4.8; IR (neat): 3479, 2960, 1709, 1641, 1252 cm-1; FAB mass: m/z 401 (M++1).
At –78°C 1.4M solution of DIBAL-H (1.07 mL, 1.5 mmol) was slowly added to a solution of α,β-unsaturated ester 44 (0.3 g, 0.75 mmol) in dry CH2Cl2 (5 mL). The solution was stirred for 2 h at –78°C before being quenched with EtOAc (5 mL). The mixture was allowed to warm to ambient temperature before an aqueous solution of Rochelle’s salt was added (30 mL) and stirred for one hour. The aqueous phase was extracted with CH2Cl2 and the combined organic extracts were dried over anhydrous Na2SO4, concentrated, and purified by column chromatography on silica gel to afford the the desired allylic alcohol 45 (0.21 g, 80%) as a pale yellow oil.; [α]D25: (+) 16.3 (c = 1.5, CHCl3); 1H NMR (CDCl3, 200MHz): δ 5.55 (d, 1H, J = 9.5), 4.01 (s, 2H), 3.70 (t, 1H, J = 2.9 Hz), 3.61 (dd, 1H, J = 10.2, 2.2 Hz), 2.4-2.6 (m, 1H), 1.78-1.70 (m, 1H), 1.68 (d, 3H, J = 1.1 Hz), 1.6 (m, 1H), 1.40-1.20 (m, 2H), 1.05 (d, 3H, J = 6.9 Hz), 0.99 (d, 3H, J = 6.9 Hz), 0.91 (s, 9H), 0.90 (t, 3H, J = 7.3 Hz), 0.73 (d, 3H, J = 6.9 Hz), 0.10 (s, 3H), 0.06 (s, 3H); 13C NMR (CDCl3, 75 MHz) : δ 134.4, 126.5, 79.9, 76.1, 68.8, 40.8, 35.5, 34.5, 28.4, 25.4, 17.6, 14.9, 13.4, 11.6, -4.6, -5.1; IR (neat): 3423, 2930, 2360, 1219, 1013 cm-1; FAB mass: m/z 359 (M++1).
Allylic alcohol 45 (0.2g, 0.55mmol) was taken up in 5 mL of CH2Cl2 and cooled to 0°C. TPP (0.365g, 1.39mmol), 2,6-lutidine (0.017mL, 0.15 mmol) and CBr4 (0.554g, 1.67mmol) were added sequentially. After 15 min, the solution was concentrated under vacuum, saturated with 10% EtOAc/hexane, and filtered through Celite. Concentration under vacuum, followed by flash chromatography through silica gel (1:9 EtOAc: Hexanes) affored allyl bromide 46 (0.218g, 93%). 1H NMR (CDCl3, 200 MHz): δ 5.69 (d, 1H, J = 9.9 Hz), 3.98 (d, 2H, J = 3.3 Hz), 3.84-3.80 (br, s, 1H), 3.69 (t, 1H, J = 2.7 Hz), 3.55 (d, 1H, J = 9.9 Hz), 2.47–2.40 (m, 1H), 1.75 (s, 3H), 1.73-1.67 (m, 1H), 1.59-1.54 (m, 1H), 1.39- 1.34 (m, 1H), 1.26-1.71 (m, 1H), 1.04 (d, 3H, J = 7.1 Hz), 0.98 (d, 3H, J = 6.6 Hz), 0.92 (s, 9H), 0.89 (t, 3H, J = 7.1 Hz), 0.721 (d, 3H, J = 6.6 Hz), 0.08 (d, 6H, J = 19.3 Hz ).
Allylic bromide 46 (0.1g, 0.23 mmol) was dissolved in dry acetonitrile (5 mL) and tributylphosphine (0.071g, 0.35 mmol) was added at once. After stirring for 30 min (or until starting material disappeared by TLC) at rt the solvent was evaporated under reduced pressure and the resulting viscous oil used directly in the next reaction.
To an ice-cooled solution of iodoxybenzoic acid (0.33 g, 1.18 mmol) in DMSO (3ml) was added a solution of alcohol 5(0.12 g, 0.78 mmol) in dry CH2Cl2 (3 mL). After stirring for 2 h at room temperature, the reaction mixture was filtered through a celite pad and washed with ether. The combined organic layers were washed with water, brine solution and dried over anhydrous Na2SO4 and concentrated invacuo. The crude product was purified by column chromatography on silica gel (EtOAc/hexane, 0.5:9.5) to give an aldehyde 47(0.083 g, 85%) as a viscous liquid; 1H NMR (CDCl3, 300 MHz): δ 9.67 (t, 1H, J = 2.0 Hz), 6.64 (dd, 1H, J = 10.4, 17.6 Hz), 5.25 (d, 1H, J = 17.6 Hz), 5.13 (t, 2H, J = 12.4 Hz), 3.24-3.18 (m, 1H), 2.36 (d, 2H, J = 8.3 Hz), 2.19 (q, 2H, J = 7.2, 15.6 Hz), 1.05 (t, 3H, J = 7.2 Hz), 1.04 (d, 3H, J = 7.2 Hz); 13C NMR (CDCl3, 75 MHz): δ 200.1, 151.3, 134.7, 129.6, 127.6, 51.0, 26.9, 21.2, 18.9, 13.2; IR (neat): 2924, 2853, 1733, 1638, 1460 cm-1; EIMS 175 (M+ + 23).
A solution of n-BuLi (1.6 M, 0.131 mL, 0.209 mmol) was added to a solution of DMSO (0.064), in dry toluene (0.55mL) at room temperature, then the whole was stirred for 45 min. A solution of phophonium salt 6 (0.090 g, 0.154 mmol) and aldehyde 47 (0.020 g, 0.080 moml) was dissolved in dry toluene (1.2 mL) was added to the solution of dimsyl carbanion at -78°C to 0°C overnight. The reaction mixture was poured into saturated aqueous NH4Cl, then the whole was extracted with Et2O. The Et2O extract was washed with saturated aqueous NaCl, then dried over MgSO4. Removal of solvent from the Et2O extract under reduced pressure gave a product, which was purified by column chromatography Et2O/ pentane 1:9) to furnish a colourless oil 3(0.67 g, 60% yield); [α]D25: (-) 20.3 (c = 0.5, CHCl3); 1H NMR (CDCl3, 200 MHz): δ 6.69-6.56 (m, 1H), 5.91 (dd, 1H, J = 11.5 Hz), 5.53-5.41 (m, 1H), 5.26-5.47 (m, 4H), 3.74-3.53 (m, 2H), 2.71-2.58 (m, 1H), 2.56-2.42 (m, 1H), 2.38-2.24 (m, 1H), 2.19 (q, 2H, J = 7.1, 14.5 Hz), 2.11-2.02 (m, 1H), 1.76 (s, 3H), 1.61-1.52 (m, 1H), 1.48-1.32 (m, 1H), 1.31-1.15 (m, 2H), 1.05 (t, 3H, J = 7.3 Hz), 0.98 (d, 3H, J = 6.7 Hz), 0.97 (d, 3H, J = 6.2 Hz), 0.88 (t, 3H, J = 7.2 Hz), 0.94-0.89 (m, 12H), 0.73 (d, 3H, J = 6.9 Hz), 0.08 (s, 3H), 0.06 (s, 3H).; 13C NMR (CDCl3, 75 MHz) : δ 141.9, 133.7, 128.6, 128.5, 127.5, 126.4, 126.0, 110.2, 80.5, 77.6, 41.4, 41.3, 35.8, 35.6, 35.4, 29.7, 29.0, 28.9, 26.2, 26.1, 25.9, 18.1, 17.9, 15.4, 13.8, 12.6, 12.1, 11.8, -4.1, -4.7; IR (neat): 3385, 2960, 2930, 1639, 1596, 1461, 1382, 1253, 1052, 1007, 836, 774 cm-1.; EIMS: m/z 499 (M++23).
To a solution of compound 2 (0.1 g, 0.806 mmol) in PhH (1 mL) was added the compound 3 (0.248 g, 1.61 mmol). The mixture was heated to 55°C, and a solution of second generation Grubbs’ catalyst (0.068 g, 0.080 mmol) in PhH (1 mL) was added via syringe pump. Heating was continued for 12 h after addition was complete. After cooling to room temperature, the mixture was filtered through a short pad of silica gel, and the filtrate was concentrated in vacuo. purification by silica gel chromatography (eluent: PE–EtOAc, 2:8) gave metathesis product dimer only. 1H NMR (CDCl3, 200 MHz): δ 6.80-6.91 (m, 2H), 6.01-6.10 (m, 4H), 4.91-5.01(m, 2H), 2.41-2.51(m, 4H).
Acknowledgement
KYG thanks UGC, New Delhi for the award of fellowship. Author acknowledges the partial support by King Saud University for Global Research Network for Organic Synthesis (GRNOS).
References
- Kobayashi M, Higuchi K, Murakami N, Tajima H, Aoki S (1997) Callystatin A, a potent cytotoxic polyketide from the marine sponge, Callyspongia truncate. Tetrahedron Lett 38(16): 2859-2862.
- Murakami N, Wang W, Aoki M, Tsutsui Y, Higuchi K, et al. (1997) Absolute stereostruc¬ture of callystatin A, a potent cytotoxic polyketide from the marine sponge, Callyspongia truncate. Tetrahedron Lett 38: 5533-5536.
- Murakami N, Wang W, Aoki M, Tsutsui Y, Sugimoto M, et al. (1998) Total synthesis of callystatin A, a potent cytotoxic imlyketide from the marine sponge, Callyspangia routtwaro. Tetrahedron Lett 39: 2349-2352.
- Murakami N, Sugimoto M, Nakajima T, Kawanishi M, Tsutsui Y, et al. (2000) Participation of the conjugated diene part for potent cytotoxicity of callystatin A, a spongean polyketide. Bioorg Med Chem 8(11): 2651-2661.
- Yadav JS, Yadagiri K, Madhuri CH, Sabitha G (2011) Studies towards the total synthesis of (−)-callystatin A. Tetrahedron Lett 52(33): 4269-4272.
- Crimmins TM, King WB (1998) Asymmetric Total Synthesis of Callystatin A: Asymmetric Aldol Additions with Titanium Enolates of Acyloxazolidinethiones. J Am Chem Soc 120: 9084-9088.
- Ammos B, Smitha BA, Benjamin M. Brandt (2001) Total Synthesis of (−)-Callystatin A. Org Lett 3: 1685-1688.
- Dias LC, Meira RRP (2002) Synthesis of C1, C11 fragment of callystatin A. Tetrahedron Lett 43(49): 8883-8885.
- Marco JL, Hueso-Rodriquez JA (1988) Synthesis of optically pure 1-(3-furyl)-1,2-dihydroxyethane Derivatives. Tetrahedron Lett 29(20): 2459-2462.
- (a) Katsuki T, Sharpless KB (1980) The first practical method for asymmetric epoxidation. J Am Chem Soc 102(18): 5974-5976. (b) Gaao Y, Hanson RM, Klunder JM, Ko SY, Masamune H, et al. (1987) Catalytic asymmetric epoxidation and kinetic resolution: modified procedures including in situ derivatization. J Am Chem Soc 109(19): 5765-5780.
- Sabitha G, Padmaja P, Sudhakar K, Yadav JS (2009) Total synthesis of the Zisomers of nonenolide and desmethyl nonenolide. Tetrahedron: Asymmetry 20(11): 1330-1336.
- Lian-Sheng L, Yu-Li W (2002) Synthesis of 3-deoxy-2-ulosonic acid KDO and 4-epi-KDN, a highly efficient approach of 3-C homologation by propargylation and oxidation. Tatrahedron 58(44): 9049-9056.
- Horita K, Yoshika T, Tanaka T, Oikawa Y, Yonemitsu O (1986) On the selectivity of deprotection of benzyl, mpm (4-methoxybenzyl) and dmpm (3,4-dimethoxybenzyl) protecting groups for hydroxy functions. Tetrahedron 42(11): 3021-3028.
- Vogel AI, Tatchell AR, Furnis BS, Hannaford AJ, Smith PWG (1996) Vogel’s text Book of practical organic chemistry. (5th edtn), Singapore publishers.871.
- Ando K (1997) Highly Selective Synthesis of Z-Unsaturated Esters by Using New Horner−Emmons Reagents, Ethyl (Diarylphosphono) acetates. J Org Chem 62(7): 1934-1939.
- Plettenburg O, Bodmer-Narkevitch V, Wong CH (2002) Synthesis of α-Galactosyl Ceramide, a Potent Immunostimulatory Agent. J Org Chem 67(13): 4559-4564.
- Yadav JS, Rao CS, Chandrasekhar S, Rao AR (1995) Asymmetric Synthesis of C19 to C27 fragment of Rifamycin-S. Tetrahedron Lett 36(42): 7717- 7720.
- a)Brown HC, Varaprasad JVN (1986) Chiral synthesis via organoboranes. 6. Hydroboration. 74. Asymmetric hydroboration of representative heterocyclicolefins with diisopinocampheylborane. Synthesis of heterocyclic boronates and heterocyclic alcohols of very high enantiomeric purity. J Am Chem Soc 108(8): 2049-2054. b) Brown HC, Desai MC, Jadav PK (1982) Hydroboration. 61. Diisopinocampheylborane of high optical purity. Improved preparation and asymmetric hydroboration of representative cis-disubstitutedalkenes. J Org Chem 47: 5065-5069.
- Corey EJ, Suggs JW (1975) Pyridinium chlorochromate. An efficient reagent for oxidation of primary and secondary alcohols to carbonyl compounds. Tetrahedron Lett 16(31): 2647-2650.
- Corey EJ, Weinshenker NM, Schaaf TK, Huber W (1969) Stereo-controlled synthesis of prostaglandins F-2a and E-2 (dl). J Am Chem Soc 91(20): 5675- 5677.
- Corey EJ, Cho H, Rucker C, Hua DH (1985) Synthesis of Amphotericin B. 2. Fragment C-D of the Aglycone. Tetrahedron Lett 26: 5239-5242.
- Mancuso AJ, Swern D (1981) Activated dimethyl sulfoxide: Useful reagents for synthesis. Synthesis 1981(3): 165-185.
- Marshall JA, Bourbeau MP (2002) Total Synthesis of (−)-Callystatin A. J Org Chem 67(9): 2751-2754.





